当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Energy Gaps of Polyradicals from an Effective and Transferable Hamiltonian with through-Bond Interactions
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-06 00:00:00 , DOI: 10.1021/acs.jctc.7b00930 Rodrigo A. Moreira 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-06 00:00:00 , DOI: 10.1021/acs.jctc.7b00930 Rodrigo A. Moreira 1
Affiliation
|
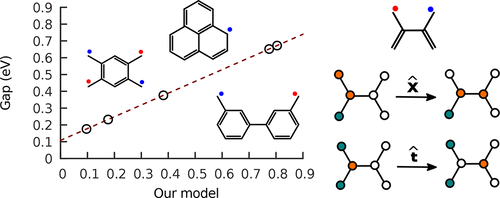
Current model Hamiltonians and ab initio many-body quantum treatments of π-conjugated polyradicals formed from hydrocarbons produce divergent results because of numerical complexity and large size of the basis-function set used. We propose an alternative, three-term Hamiltonian, to describe these various polyradicals that simplifies considerably the computational cost while providing a physical interpretation for all three terms and a high degree of model universality. The essential feature of this Hamiltonian is a term, not present in previous models, describing the three-sited through-bond interaction that governs the noninteracting spin-up and spin-down sectors. A computation of the lowest energy gaps and spin configurations for the smaller polyradicals demonstrates the efficacy of the model and its potential in applications in revealing electrical conductivity and ferromagnetism of the more complicated substituted polyradicals.
中文翻译:

来自全键相互作用的有效可转移哈密顿量的多自由基能隙
当前模型哈密顿量和从头算由碳氢化合物形成的π共轭多自由基的多体量子处理由于数值复杂性和所使用的基函数集的大小而产生不同的结果。我们提出了一种替代的三项哈密顿量,以描述这些各种多基自由基,从而大大简化了计算成本,同时提供了对所有三项的物理解释和高度的模型通用性。该哈密顿量的基本特征是一个术语,该术语在以前的模型中不存在,它描述了控制非相互作用的上旋和下旋扇区的三位键间相互作用。
更新日期:2017-12-06
中文翻译:
来自全键相互作用的有效可转移哈密顿量的多自由基能隙
当前模型哈密顿量和从头算由碳氢化合物形成的π共轭多自由基的多体量子处理由于数值复杂性和所使用的基函数集的大小而产生不同的结果。我们提出了一种替代的三项哈密顿量,以描述这些各种多基自由基,从而大大简化了计算成本,同时提供了对所有三项的物理解释和高度的模型通用性。该哈密顿量的基本特征是一个术语,该术语在以前的模型中不存在,它描述了控制非相互作用的上旋和下旋扇区的三位键间相互作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号