当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Macrocycle Conformational Sampling by DFT-D3/COSMO-RS Methodology
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2017-12-18 00:00:00 , DOI: 10.1021/acs.jcim.7b00453 Ondrej Gutten 1 , Daniel Bím 1 , Jan Řezáč 1 , Lubomír Rulíšek 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2017-12-18 00:00:00 , DOI: 10.1021/acs.jcim.7b00453 Ondrej Gutten 1 , Daniel Bím 1 , Jan Řezáč 1 , Lubomír Rulíšek 1
Affiliation
|
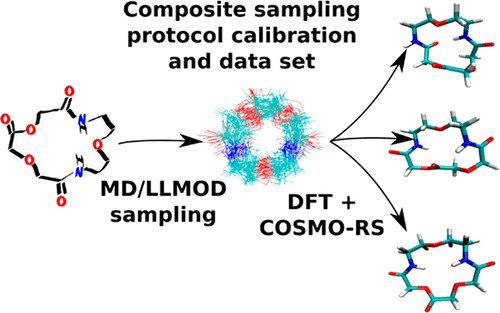
To find and calibrate a robust and reliable computational protocol for mapping conformational space of medium-sized molecules, exhaustive conformational sampling has been carried out for a series of seven macrocyclic compounds of varying ring size and one acyclic analogue. While five of them were taken from the MD/LLMOD/force field study by Shelley and co-workers (Watts, K. S.; Dalal, P.; Tebben, A. J.; Cheney, D. L.; Shelley, J. C. Macrocycle Conformational Sampling with MacroModel. J. Chem. Inf. Model. 2014, 54, 2680−2696), three represent potential macrocyclic inhibitors of human cyclophilin A. The free energy values (GDFT/COSMO-RS) for all of the conformers of each compound were obtained by a composite protocol based on in vacuo quantum mechanics (DFT-D3 method in a large basis set), standard gas-phase thermodynamics, and the COSMO-RS solvation model. The GDFT/COSMO-RS values were used as the reference for evaluating the performance of conformational sampling algorithms: standard and extended MD/LLMOD search (simulated-annealing molecular dynamics with low-lying eigenvector following algorithms, employing the OPLS2005 force field plus GBSA solvation) available in MacroModel and plain molecular dynamics (MD) sampling at high temperature (1000 K) using the semiempirical quantum mechanics (SQM) potential SQM(PM6-D3H4/COSMO) followed by energy minimization of the snapshots. It has been shown that the former protocol (MD/LLMOD) may provide a more complete set of initial structures that ultimately leads to the identification of a greater number of low-energy conformers (as assessed by GDFT/COSMO-RS) than the latter (i.e., plain SQM MD). The CPU time needed to fully evaluate one medium-sized compound (∼100 atoms, typically resulting in several hundred or a few thousand conformers generated and treated quantum-mechanically) is approximately 1,000–100,000 CPU hours on today’s computers, which transforms to 1–7 days on a small-sized computer cluster with a few hundred CPUs. Finally, our data sets based on the rigorous quantum-chemical approach allow us to formulate a composite conformational sampling protocol with multiple checkpoints and truncation of redundant structural data that offers superior insights at affordable computational cost.
中文翻译:

DFT-D3 / COSMO-RS方法进行大周期构象采样
为了找到并校准用于映射中型分子构象空间的可靠可靠的计算方案,已经对一系列七个不同环大小的大环化合物和一个无环类似物进行了详尽的构象采样。其中有五个是由雪莱及其同事从MD / LLMOD /部队实地研究中提取的(堪萨斯州瓦茨 Dalal,P .; AJ Tebben;切尼(DL);雪莱(JC) 使用MacroModel进行Macrocycle构象采样。J.化学。Inf。模型。 2014,54,2680至2696年),三是人类亲环蛋白A的电位大环抑制剂自由能的值(g ^ DFT / COSMO-RS对所有各化合物的构象异构体)用复合协议基于所获得真空量子力学(大型基础上的DFT-D3方法),标准气相热力学和COSMO-RS溶剂化模型。所述ģ DFT / COSMO-RS值用作评估构象采样算法性能的参考:MacroModel和平原提供了标准和扩展的MD / LLMOD搜索(采用低特征向量的模拟退火分子动力学,遵循以下算法,采用OPLS2005力场加GBSA溶剂化)使用半经验量子力学(SQM)势SQM(PM6-D3H4 / COSMO)在高温(1000 K)下进行分子动力学(MD)采样,然后将快照的能量最小化。已经证明,前一种协议(MD / LLMOD)可以提供更完整的初始结构集,最终导致鉴定出更多的低能构象体(如G DFT / COSMO-RS所评估)),而不是后者(即普通的SQM MD)。在当今的计算机上,要完全评估一种中等大小的化合物(约100个原子,通常会产生数百个或几千个构象异构体并进行量子力学处理),其CPU时间约为1,000–100,000 CPU小时。在具有几百个CPU的小型计算机群集上运行7天。最后,基于严格的量子化学方法的数据集使我们能够制定具有多个检查点的复合构象采样协议,并截短冗余结构数据,从而以可承受的计算成本提供卓越的见解。
更新日期:2017-12-18
中文翻译:
DFT-D3 / COSMO-RS方法进行大周期构象采样
为了找到并校准用于映射中型分子构象空间的可靠可靠的计算方案,已经对一系列七个不同环大小的大环化合物和一个无环类似物进行了详尽的构象采样。其中有五个是由雪莱及其同事从MD / LLMOD /部队实地研究中提取的(










































 京公网安备 11010802027423号
京公网安备 11010802027423号