当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Comparison between the Bethe–Salpeter Equation and Configuration Interaction Approaches for Solving a Quantum Chemistry Problem: Calculating the Excitation Energy for Finite 1D Hubbard Chains
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-01-08 00:00:00 , DOI: 10.1021/acs.jctc.7b00246 Qi Ou 1 , Joseph E. Subotnik 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-01-08 00:00:00 , DOI: 10.1021/acs.jctc.7b00246 Qi Ou 1 , Joseph E. Subotnik 1
Affiliation
|
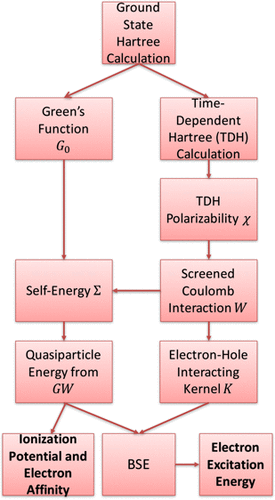
We calculate the excitation energies of finite 1D Hubbard chains with a variety of different site energies from two perspectives: (i) the physics-based Bethe–Salpeter equation (BSE) method and (ii) the chemistry-based configuration interaction (CI) approach. Results obtained from all methods are compared against the exact values for three classes of systems: metallic, impurity-doped, and molecular (semiconducting/insulating) systems. While in a previous study we showed that the GW method holds comparative advantages versus traditional quantum chemistry approaches for calculating the ionization potentials and electron affinities across a large range of Hamiltonians, we show now that the BSE method outperforms CI approaches only for metallic and semiconducting systems. For insulating molecular systems, CI approaches generate better results.
中文翻译:

解决量子化学问题的Bethe–Salpeter方程和构型相互作用方法之间的比较:计算有限一维Hubbard链的激发能
我们从两个角度计算具有各种不同站点能量的有限一维Hubbard链的激发能:(i)基于物理学的Bethe-Salpeter方程(BSE)方法和(ii)基于化学构型相互作用(CI)的方法。将所有方法获得的结果与三类系统的精确值进行比较:金属,掺杂系统和分子(半导体/绝缘)系统。在先前的研究中,我们发现GW与传统的量子化学方法相比,该方法具有比较优势,可以在很大的哈密顿量范围内计算电离势和电子亲和力,现在我们证明,BSE方法仅在金属和半导体系统中优于CI方法。对于绝缘分子系统,CI方法可产生更好的结果。
更新日期:2018-01-08
中文翻译:
解决量子化学问题的Bethe–Salpeter方程和构型相互作用方法之间的比较:计算有限一维Hubbard链的激发能
我们从两个角度计算具有各种不同站点能量的有限一维Hubbard链的激发能:(i)基于物理学的Bethe-Salpeter方程(BSE)方法和(ii)基于化学构型相互作用(CI)的方法。将所有方法获得的结果与三类系统的精确值进行比较:金属,掺杂系统和分子(半导体/绝缘)系统。在先前的研究中,我们发现GW与传统的量子化学方法相比,该方法具有比较优势,可以在很大的哈密顿量范围内计算电离势和电子亲和力,现在我们证明,BSE方法仅在金属和半导体系统中优于CI方法。对于绝缘分子系统,CI方法可产生更好的结果。










































 京公网安备 11010802027423号
京公网安备 11010802027423号