当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Can MM/GBSA calculations be sped up by system truncation?
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2017-11-26 , DOI: 10.1002/jcc.25120 Majda Misini Ignjatović 1 , Paulius Mikulskis 1 , Pär Söderhjelm 2 , Ulf Ryde 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2017-11-26 , DOI: 10.1002/jcc.25120 Majda Misini Ignjatović 1 , Paulius Mikulskis 1 , Pär Söderhjelm 2 , Ulf Ryde 1
Affiliation
|
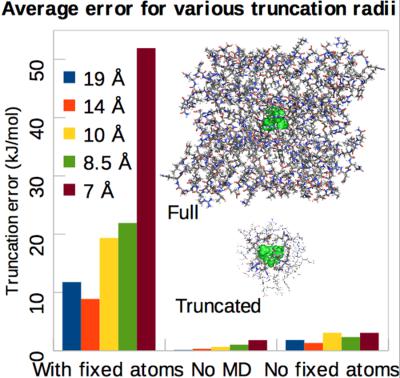
We have studied whether calculations of the binding free energy of small ligands to a protein by the MM/GBSA approach (molecular mechanics combined with generalized Born and surface area solvation) can be sped up by including only a restricted number of atoms close to the ligand. If the protein is truncated before the molecular dynamics (MD) simulations, quite large changes are observed for the calculated binding energies, for example, 4 kJ/mol average difference for a radius of 19 Å for the binding of nine phenol derivatives to ferritin. The results are improved if no atoms are fixed in the simulations, with average and maximum errors of 2 and 3 kJ/mol at 19 Å and 3 and 6 kJ/mol at 7 Å. Similar results are obtained for two additional proteins, p38α MAP kinase and factor Xa. On the other hand, if energies are calculated on snapshots that are truncated after the MD simulation, all residues more than 8.5 Å from the ligand can be omitted without changing the energies by more than 1 kJ/mol on average (maximum error 1.4 kJ/mol). At the molecular mechanics level, the gain in computer time for such an approach is small. However, it shows what size of system should be used if the energies instead are calculated with a more demanding method, for example, quantum‐mechanics. © 2017 Wiley Periodicals, Inc.
中文翻译:

MM/GBSA 计算可以通过系统截断来加速吗?
我们研究了通过 MM/GBSA 方法(分子力学结合广义玻恩和表面积溶剂化)计算小配体与蛋白质的结合自由能是否可以通过仅包含有限数量的靠近配体的原子来加速. 如果蛋白质在分子动力学 (MD) 模拟之前被截断,则会观察到计算结合能的相当大的变化,例如,对于 9 种苯酚衍生物与铁蛋白的结合,半径为 19 Å 的平均差异为 4 kJ/mol。如果模拟中没有固定原子,结果会得到改善,平均和最大误差在 19 Å 时为 2 和 3 kJ/mol,在 7 Å 时为 3 和 6 kJ/mol。对于另外两种蛋白质,p38α MAP 激酶和因子 Xa,也获得了类似的结果。另一方面,如果在 MD 模拟后截断的快照上计算能量,则可以省略所有距离配体超过 8.5 Å 的残基,而平均能量变化不超过 1 kJ/mol(最大误差为 1.4 kJ/mol)。在分子力学水平上,这种方法在计算机时间上的增益很小。但是,它显示了如果使用更苛刻的方法(例如量子力学)来计算能量,则应该使用多大的系统。© 2017 威利期刊公司。它显示了如果使用更苛刻的方法(例如量子力学)来计算能量,则应该使用多大的系统。© 2017 威利期刊公司。它显示了如果使用更苛刻的方法(例如量子力学)来计算能量,则应该使用多大的系统。© 2017 威利期刊公司。
更新日期:2017-11-26
中文翻译:
MM/GBSA 计算可以通过系统截断来加速吗?
我们研究了通过 MM/GBSA 方法(分子力学结合广义玻恩和表面积溶剂化)计算小配体与蛋白质的结合自由能是否可以通过仅包含有限数量的靠近配体的原子来加速. 如果蛋白质在分子动力学 (MD) 模拟之前被截断,则会观察到计算结合能的相当大的变化,例如,对于 9 种苯酚衍生物与铁蛋白的结合,半径为 19 Å 的平均差异为 4 kJ/mol。如果模拟中没有固定原子,结果会得到改善,平均和最大误差在 19 Å 时为 2 和 3 kJ/mol,在 7 Å 时为 3 和 6 kJ/mol。对于另外两种蛋白质,p38α MAP 激酶和因子 Xa,也获得了类似的结果。另一方面,如果在 MD 模拟后截断的快照上计算能量,则可以省略所有距离配体超过 8.5 Å 的残基,而平均能量变化不超过 1 kJ/mol(最大误差为 1.4 kJ/mol)。在分子力学水平上,这种方法在计算机时间上的增益很小。但是,它显示了如果使用更苛刻的方法(例如量子力学)来计算能量,则应该使用多大的系统。© 2017 威利期刊公司。它显示了如果使用更苛刻的方法(例如量子力学)来计算能量,则应该使用多大的系统。© 2017 威利期刊公司。它显示了如果使用更苛刻的方法(例如量子力学)来计算能量,则应该使用多大的系统。© 2017 威利期刊公司。










































 京公网安备 11010802027423号
京公网安备 11010802027423号