当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mechanistic insights on ethanol dehydrogenation on Pd–Au model catalysts: a combined experimental and DFT study
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2017-11-02 00:00:00 , DOI: 10.1039/c7cp05097f E. J. Evans 1, 2, 3, 4, 5 , H. Li 3, 6, 7, 8, 9 , Wen-Yueh Yu 1, 2, 3, 4, 5 , G. M. Mullen 1, 2, 3, 4, 5 , G. Henkelman 3, 6, 7, 8, 9 , C. Buddie Mullins 1, 2, 3, 4, 5
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2017-11-02 00:00:00 , DOI: 10.1039/c7cp05097f E. J. Evans 1, 2, 3, 4, 5 , H. Li 3, 6, 7, 8, 9 , Wen-Yueh Yu 1, 2, 3, 4, 5 , G. M. Mullen 1, 2, 3, 4, 5 , G. Henkelman 3, 6, 7, 8, 9 , C. Buddie Mullins 1, 2, 3, 4, 5
Affiliation
|
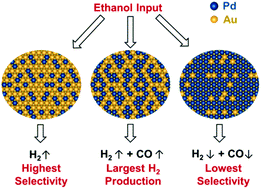
In this study, we have combined ultra-high vacuum (UHV) experiments and density functional theory (DFT) calculations to investigate ethanol (EtOH) dehydrogenation on Pd–Au model catalysts. Using EtOH reactive molecular beam scattering (RMBS), EtOH temperature-programmed desorption (TPD), and DFT calculations, we show how different Pd ensemble sizes on Au(111) can affect the mechanism for EtOH dehydrogenation and H2 production. The Au(111) surface with an initial coverage of 2 monolayers of Pd (2 ML Pd–Au) had the highest H2 yield. However, the 1 ML Pd–Au catalyst showed the highest selectivity and stability, yielding appreciable amounts of only H2 and acetaldehyde. Arrhenius plots of H2 production confirm that the mechanisms for EtOH dehydrogenation differed between 1 and 2 ML Pd–Au, supporting the perceived difference in selectivity between the two surfaces. DFT calculations support this difference in mechanism, showing a dependence of the initial dehydrogenation selectivity of EtOH on the size of Pd ensemble. DFT binding energies and EtOH TPD confirm that EtOH has increasing surface affinity with increasing Pd ensemble size and Pd coverage, indicating that surfaces with more Pd are more likely to induce an EtOH reaction instead of desorb. Our theoretical results show that the synergistic influence of atomic ensemble and electronic effects on Pd/Au(111) can lead to different H2 association energies and EtOH dehydrogenation capacities at different Pd ensembles. These results provide mechanistic insights into ethanol's dehydrogenation interactions with different sites on the Pd–Au surface and can potentially aid in bimetallic catalyst design for applications such as fuel cells.
中文翻译:

在Pd-Au模型催化剂上进行乙醇脱氢的机理见解:结合实验和DFT研究
在这项研究中,我们结合了超高真空(UHV)实验和密度泛函理论(DFT)计算,以研究Pd–Au模型催化剂上的乙醇(EtOH)脱氢。使用EtOH反应性分子束散射(RMBS),EtOH程序升温解吸(TPD)和DFT计算,我们显示了Au(111)上不同的Pd集合尺寸如何影响EtOH脱氢和H 2产生的机理。最初覆盖2个单层Pd(2 ML Pd–Au)的Au(111)表面具有最高的H 2收率。但是,1 ML Pd–Au催化剂显示出最高的选择性和稳定性,仅产生相当数量的H 2和乙醛。H 2的阿累尼乌斯情节生产证实,EtOH脱氢的机理在1和2 ML Pd–Au之间有所不同,这支持了两个表面之间选择性的感知差异。DFT计算支持了这种机理上的差异,表明EtOH的初始脱氢选择性与Pd团簇的大小有关。DFT结合能和EtOH TPD证实EtOH具有随着Pd团簇尺寸和Pd覆盖率增加而增加的表面亲和力,表明具有更多Pd的表面更有可能引发EtOH反应而不是解吸。我们的理论结果表明,原子团簇和电子效应对Pd / Au(111)的协同影响可以导致不同的H 2。Pd集成时的缔合能和EtOH脱氢能力。这些结果为深入了解乙醇与Pd-Au表面不同位置的脱氢相互作用提供了机械依据,并有可能有助于燃料电池等应用的双金属催化剂设计。
更新日期:2017-11-22
中文翻译:
在Pd-Au模型催化剂上进行乙醇脱氢的机理见解:结合实验和DFT研究
在这项研究中,我们结合了超高真空(UHV)实验和密度泛函理论(DFT)计算,以研究Pd–Au模型催化剂上的乙醇(EtOH)脱氢。使用EtOH反应性分子束散射(RMBS),EtOH程序升温解吸(TPD)和DFT计算,我们显示了Au(111)上不同的Pd集合尺寸如何影响EtOH脱氢和H 2产生的机理。最初覆盖2个单层Pd(2 ML Pd–Au)的Au(111)表面具有最高的H 2收率。但是,1 ML Pd–Au催化剂显示出最高的选择性和稳定性,仅产生相当数量的H 2和乙醛。H 2的阿累尼乌斯情节生产证实,EtOH脱氢的机理在1和2 ML Pd–Au之间有所不同,这支持了两个表面之间选择性的感知差异。DFT计算支持了这种机理上的差异,表明EtOH的初始脱氢选择性与Pd团簇的大小有关。DFT结合能和EtOH TPD证实EtOH具有随着Pd团簇尺寸和Pd覆盖率增加而增加的表面亲和力,表明具有更多Pd的表面更有可能引发EtOH反应而不是解吸。我们的理论结果表明,原子团簇和电子效应对Pd / Au(111)的协同影响可以导致不同的H 2。Pd集成时的缔合能和EtOH脱氢能力。这些结果为深入了解乙醇与Pd-Au表面不同位置的脱氢相互作用提供了机械依据,并有可能有助于燃料电池等应用的双金属催化剂设计。










































 京公网安备 11010802027423号
京公网安备 11010802027423号