当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Reaction kinetics of hydrogen abstraction from polycyclic aromatic hydrocarbons by H atoms
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2017-10-31 00:00:00 , DOI: 10.1039/c7cp04964a Dingyu Hou 1, 2, 3, 4, 5 , Xiaoqing You 1, 2, 3, 4, 5
Physical Chemistry Chemical Physics ( IF 3.3 ) Pub Date : 2017-10-31 00:00:00 , DOI: 10.1039/c7cp04964a Dingyu Hou 1, 2, 3, 4, 5 , Xiaoqing You 1, 2, 3, 4, 5
Affiliation
|
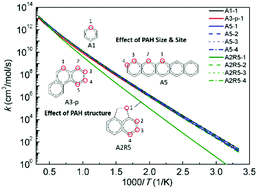
Hydrogen abstraction reactions of polycyclic aromatic hydrocarbons (PAH) by H atoms play a very important role in both PAH and soot formation processes. However, large discrepancies up to a few orders of magnitude exist among the literature rate constant values. To increase the reliability of the computed rate constants, it is critical to obtain highly accurate potential energy surfaces. For this purpose, we have investigated the energetics of hydrogen abstraction from benzene and naphthalene using both high level-of-theory quantum chemistry methods and a series of density functional theory (DFT) methods, among which M06-2X/6-311g(d,p) has the best performance with a mean unsigned deviation from the CCSD(T)/CBS calculations of 1.0 kcal mol−1 for barrier heights and reaction energies. Thus, M06-2X/6-311g(d,p) has then been applied to compute the potential energy surfaces of the hydrogen abstraction reactions of a series of larger PAH. Based on the quantum chemistry calculations, rate constants are computed using the canonical transition state theory. The effects of the PAH size, structure, and reaction site on the energetics and rate constants are examined systematically. Finally, the hydrogen abstraction rate constants for application in PAH and soot surface chemistry models are recommended.
中文翻译:

H原子从多环芳烃中析氢的反应动力学
H原子引起的多环芳烃(PAH)的氢提取反应在PAH和烟灰形成过程中都起着非常重要的作用。但是,文献速率常数之间存在高达几个数量级的巨大差异。为了提高计算出的速率常数的可靠性,获取高度精确的势能面至关重要。为此,我们使用高水平的理论量子化学方法和一系列的密度泛函理论(DFT)方法研究了苯和萘中氢的提取能量,其中M06-2X / 6-311g(d ,p)具有最佳性能,与CCSD(T)/ CBS计算的平均无符号偏差为1.0 kcal mol -1用于势垒高度和反应能。因此,随后已将M06-2X / 6-311g(d,p)用于计算一系列较大的PAH的氢提取反应的势能面。基于量子化学计算,使用标准跃迁状态理论计算速率常数。系统地检查了PAH的大小,结构和反应部位对能量和速率常数的影响。最后,推荐用于PAH和烟灰表面化学模型的氢提取速率常数。
更新日期:2017-11-22
中文翻译:
H原子从多环芳烃中析氢的反应动力学
H原子引起的多环芳烃(PAH)的氢提取反应在PAH和烟灰形成过程中都起着非常重要的作用。但是,文献速率常数之间存在高达几个数量级的巨大差异。为了提高计算出的速率常数的可靠性,获取高度精确的势能面至关重要。为此,我们使用高水平的理论量子化学方法和一系列的密度泛函理论(DFT)方法研究了苯和萘中氢的提取能量,其中M06-2X / 6-311g(d ,p)具有最佳性能,与CCSD(T)/ CBS计算的平均无符号偏差为1.0 kcal mol -1用于势垒高度和反应能。因此,随后已将M06-2X / 6-311g(d,p)用于计算一系列较大的PAH的氢提取反应的势能面。基于量子化学计算,使用标准跃迁状态理论计算速率常数。系统地检查了PAH的大小,结构和反应部位对能量和速率常数的影响。最后,推荐用于PAH和烟灰表面化学模型的氢提取速率常数。


























 京公网安备 11010802027423号
京公网安备 11010802027423号