当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Analytic energy gradients for orbital-optimized MP3 and MP2.5 with the density-fitting approximation: An efficient implementation
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2017-11-21 , DOI: 10.1002/jcc.25122 Uğur Bozkaya 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2017-11-21 , DOI: 10.1002/jcc.25122 Uğur Bozkaya 1
Affiliation
|
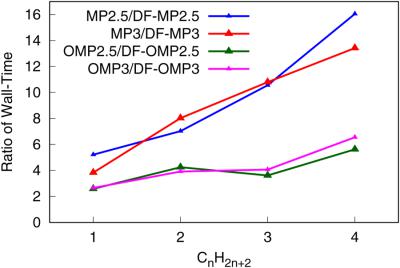
Efficient implementations of analytic gradients for the orbital‐optimized MP3 and MP2.5 and their standard versions with the density‐fitting approximation, which are denoted as DF‐MP3, DF‐MP2.5, DF‐OMP3, and DF‐OMP2.5, are presented. The DF‐MP3, DF‐MP2.5, DF‐OMP3, and DF‐OMP2.5 methods are applied to a set of alkanes and noncovalent interaction complexes to compare the computational cost with the conventional MP3, MP2.5, OMP3, and OMP2.5. Our results demonstrate that density‐fitted perturbation theory (DF‐MP) methods considered substantially reduce the computational cost compared to conventional MP methods. The efficiency of our DF‐MP methods arise from the reduced input/output (I/O) time and the acceleration of gradient related terms, such as computations of particle density and generalized Fock matrices (PDMs and GFM), solution of the Z‐vector equation, back‐transformations of PDMs and GFM, and evaluation of analytic gradients in the atomic orbital basis. Further, application results show that errors introduced by the DF approach are negligible. Mean absolute errors for bond lengths of a molecular set, with the cc‐pCVQZ basis set, is 0.0001–0.0002 Å. © 2017 Wiley Periodicals, Inc.
中文翻译:

具有密度拟合近似的轨道优化 MP3 和 MP2.5 的解析能量梯度:一种有效的实现
轨道优化的 MP3 和 MP2.5 及其标准版本的解析梯度的有效实现与密度拟合近似,表示为 DF-MP3、DF-MP2.5、DF-OMP3 和 DF-OMP2.5 , 呈现。将 DF-MP3、DF-MP2.5、DF-OMP3 和 DF-OMP2.5 方法应用于一组烷烃和非共价相互作用复合物,以将计算成本与常规 MP3、MP2.5、OMP3 和OMP2.5。我们的结果表明,与传统的 MP 方法相比,密度拟合微扰理论 (DF-MP) 方法大大降低了计算成本。我们的 DF-MP 方法的效率源于输入/输出 (I/O) 时间的减少和梯度相关项的加速,例如粒子密度和广义 Fock 矩阵(PDM 和 GFM)的计算,Z 向量方程的解,PDM 和 GFM 的反变换,以及原子轨道基础中解析梯度的评估。此外,应用结果表明DF方法引入的误差可以忽略不计。具有 cc-pCVQZ 基组的分子组键长的平均绝对误差为 0.0001–0.0002 Å。© 2017 威利期刊公司。
更新日期:2017-11-21
中文翻译:
具有密度拟合近似的轨道优化 MP3 和 MP2.5 的解析能量梯度:一种有效的实现
轨道优化的 MP3 和 MP2.5 及其标准版本的解析梯度的有效实现与密度拟合近似,表示为 DF-MP3、DF-MP2.5、DF-OMP3 和 DF-OMP2.5 , 呈现。将 DF-MP3、DF-MP2.5、DF-OMP3 和 DF-OMP2.5 方法应用于一组烷烃和非共价相互作用复合物,以将计算成本与常规 MP3、MP2.5、OMP3 和OMP2.5。我们的结果表明,与传统的 MP 方法相比,密度拟合微扰理论 (DF-MP) 方法大大降低了计算成本。我们的 DF-MP 方法的效率源于输入/输出 (I/O) 时间的减少和梯度相关项的加速,例如粒子密度和广义 Fock 矩阵(PDM 和 GFM)的计算,Z 向量方程的解,PDM 和 GFM 的反变换,以及原子轨道基础中解析梯度的评估。此外,应用结果表明DF方法引入的误差可以忽略不计。具有 cc-pCVQZ 基组的分子组键长的平均绝对误差为 0.0001–0.0002 Å。© 2017 威利期刊公司。










































 京公网安备 11010802027423号
京公网安备 11010802027423号