当前位置:
X-MOL 学术
›
Chem. Bio. Drug Des.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Characterization of the interaction forces in a drug carrier complex of doxorubicin with a drug‐binding peptide
Chemical Biology & Drug Design ( IF 3.2 ) Pub Date : 2017-12-21 , DOI: 10.1111/cbdd.13151 Gergana Gocheva 1 , Nina Ilieva 1 , Kalina Peneva 2 , Anela Ivanova 1
Chemical Biology & Drug Design ( IF 3.2 ) Pub Date : 2017-12-21 , DOI: 10.1111/cbdd.13151 Gergana Gocheva 1 , Nina Ilieva 1 , Kalina Peneva 2 , Anela Ivanova 1
Affiliation
|
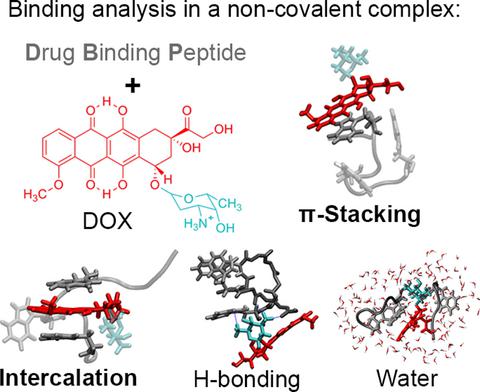
Polypeptide‐based materials are used as building blocks for drug delivery systems aimed at toxicity decrease in chemotherapeutics. A molecular‐level approach is adopted for investigating the non‐covalent interactions between doxorubicin and a recently synthesized drug‐binging peptide as a key part of a system for delivery to neoplastic cells. Molecular dynamics simulations in aqueous solution at room and body temperature are applied to investigate the structure and the binding modes within the drug–peptide complex. The tryptophans are outlined as the main chemotherapeutic adsorption sites, and the importance of their placement in the peptide sequence is highlighted. The drug–peptide binging energy is evaluated by density functional theory calculations. Principal component analysis reveals comparable importance of several types of interaction for the binding strength. π‐Stacking is dominant, but other factors are also significant: intercalation, peptide backbone stacking, electrostatics, dispersion, and solvation. Intra‐ and intermolecular H‐bonding also stabilizes the complexes. The influence of solvent molecules on the binding energy is mild. The obtained data characterize the drug‐to‐peptide attachment as a mainly attractive collective process with interactions spanning a broad range of values. These results explain with atomistic detail the experimentally registered doxorubicin‐binging ability of the peptide and outline the complex as a prospective carrying unit that can be employed in design of drug delivery systems.
中文翻译:

阿霉素与药物结合肽的药物载体复合物中相互作用力的表征
基于多肽的材料用作药物递送系统的组成部分,旨在降低化疗药物的毒性。采用分子水平的方法研究阿霉素与最近合成的药物结合肽之间的非共价相互作用,这是传递给肿瘤细胞的系统的关键部分。在室温和室温下在水溶液中进行分子动力学模拟,以研究药物-肽复合物中的结构和结合模式。色氨酸被概述为主要的化学吸附位点,突出了它们在肽序列中的重要性。药物-肽结合能通过密度泛函理论计算来评估。主成分分析表明,几种类型的相互作用对于结合强度具有相当的重要性。π-堆积是主要因素,但其他因素也很重要:插层,肽主链堆积,静电,分散和溶剂化。分子内和分子间的氢键也稳定了络合物。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。但其他因素也很重要:插层,肽主链堆积,静电,分散和溶剂化。分子内和分子间的氢键也稳定了络合物。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。但其他因素也很重要:插层,肽主链堆积,静电,分散和溶剂化。分子内和分子间的氢键也稳定了络合物。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。
更新日期:2017-12-21
中文翻译:
阿霉素与药物结合肽的药物载体复合物中相互作用力的表征
基于多肽的材料用作药物递送系统的组成部分,旨在降低化疗药物的毒性。采用分子水平的方法研究阿霉素与最近合成的药物结合肽之间的非共价相互作用,这是传递给肿瘤细胞的系统的关键部分。在室温和室温下在水溶液中进行分子动力学模拟,以研究药物-肽复合物中的结构和结合模式。色氨酸被概述为主要的化学吸附位点,突出了它们在肽序列中的重要性。药物-肽结合能通过密度泛函理论计算来评估。主成分分析表明,几种类型的相互作用对于结合强度具有相当的重要性。π-堆积是主要因素,但其他因素也很重要:插层,肽主链堆积,静电,分散和溶剂化。分子内和分子间的氢键也稳定了络合物。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。但其他因素也很重要:插层,肽主链堆积,静电,分散和溶剂化。分子内和分子间的氢键也稳定了络合物。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。但其他因素也很重要:插层,肽主链堆积,静电,分散和溶剂化。分子内和分子间的氢键也稳定了络合物。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。溶剂分子对结合能的影响很小。所获得的数据将药物与肽的结合表征为主要有吸引力的集体过程,相互作用范围广泛。这些结果用原子学的细节解释了该肽在实验上具有的阿霉素结合能力,并概述了该复合物作为可用于药物输送系统设计的预期携带单位。










































 京公网安备 11010802027423号
京公网安备 11010802027423号