当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Comparison of Quantitative Mass Spectrometry Platforms for Monitoring Kinase ATP Probe Uptake in Lung Cancer
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2017-11-22 00:00:00 , DOI: 10.1021/acs.jproteome.7b00329 Melissa A. Hoffman 1, 2 , Bin Fang 1 , Eric B. Haura 1 , Uwe Rix 1 , John M. Koomen 1
Journal of Proteome Research ( IF 3.8 ) Pub Date : 2017-11-22 00:00:00 , DOI: 10.1021/acs.jproteome.7b00329 Melissa A. Hoffman 1, 2 , Bin Fang 1 , Eric B. Haura 1 , Uwe Rix 1 , John M. Koomen 1
Affiliation
|
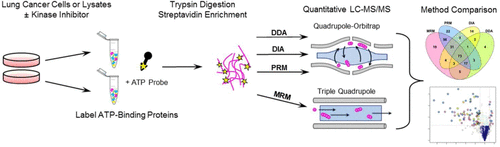
Recent developments in instrumentation and bioinformatics have led to new quantitative mass spectrometry platforms including LC–MS/MS with data-independent acquisition (DIA) and targeted analysis using parallel reaction monitoring mass spectrometry (LC–PRM), which provide alternatives to well-established methods, such as LC–MS/MS with data-dependent acquisition (DDA) and targeted analysis using multiple reaction monitoring mass spectrometry (LC–MRM). These tools have been used to identify signaling perturbations in lung cancers and other malignancies, supporting the development of effective kinase inhibitors and, more recently, providing insights into therapeutic resistance mechanisms and drug repurposing opportunities. However, detection of kinases in biological matrices can be challenging; therefore, activity-based protein profiling enrichment of ATP-utilizing proteins was selected as a test case for exploring the limits of detection of low-abundance analytes in complex biological samples. To examine the impact of different MS acquisition platforms, quantification of kinase ATP uptake following kinase inhibitor treatment was analyzed by four different methods: LC–MS/MS with DDA and DIA, LC–MRM, and LC–PRM. For discovery data sets, DIA increased the number of identified kinases by 21% and reduced missingness when compared with DDA. In this context, MRM and PRM were most effective at identifying global kinome responses to inhibitor treatment, highlighting the value of a priori target identification and manual evaluation of quantitative proteomics data sets. We compare results for a selected set of desthiobiotinylated peptides from PRM, MRM, and DIA and identify considerations for selecting a quantification method and postprocessing steps that should be used for each data acquisition strategy.
中文翻译:

监测肺癌激酶ATP探针摄取的定量质谱平台的比较
仪器和生物信息学的最新发展催生了新的定量质谱分析平台,包括具有数据独立采集(DIA)的LC–MS / MS和使用平行反应监测质谱(LC–PRM)的靶向分析,这为完善的方法提供了替代方案方法,例如具有数据依赖采集(DDA)的LC–MS / MS和使用多反应监测质谱(LC–MRM)的靶向分析。这些工具已被用于识别肺癌和其他恶性肿瘤中的信号扰动,支持有效激酶抑制剂的开发,并且最近提供了对治疗耐药性机制和药物再利用机会的见解。然而,在生物基质中检测激酶可能具有挑战性。所以,选择基于活性的基于ATP的蛋白质的蛋白质谱分析作为探索复杂生物样品中低丰度分析物检测极限的测试案例。为了检查不同MS采集平台的影响,通过四种不同的方法对激酶抑制剂处理后激酶ATP摄取的定量进行了分析:带有DDA和DIA的LC-MS / MS,LC-MRM和LC-PRM。对于发现数据集,与DDA相比,DIA将鉴定出的激酶数量增加了21%,并减少了缺失。在这种情况下,MRM和PRM最有效地确定了对抑制剂治疗的整体运动学反应,突显了先验靶点鉴定和定量蛋白质组学数据集手动评估的价值。我们比较了从PRM,MRM,
更新日期:2017-11-22
中文翻译:
监测肺癌激酶ATP探针摄取的定量质谱平台的比较
仪器和生物信息学的最新发展催生了新的定量质谱分析平台,包括具有数据独立采集(DIA)的LC–MS / MS和使用平行反应监测质谱(LC–PRM)的靶向分析,这为完善的方法提供了替代方案方法,例如具有数据依赖采集(DDA)的LC–MS / MS和使用多反应监测质谱(LC–MRM)的靶向分析。这些工具已被用于识别肺癌和其他恶性肿瘤中的信号扰动,支持有效激酶抑制剂的开发,并且最近提供了对治疗耐药性机制和药物再利用机会的见解。然而,在生物基质中检测激酶可能具有挑战性。所以,选择基于活性的基于ATP的蛋白质的蛋白质谱分析作为探索复杂生物样品中低丰度分析物检测极限的测试案例。为了检查不同MS采集平台的影响,通过四种不同的方法对激酶抑制剂处理后激酶ATP摄取的定量进行了分析:带有DDA和DIA的LC-MS / MS,LC-MRM和LC-PRM。对于发现数据集,与DDA相比,DIA将鉴定出的激酶数量增加了21%,并减少了缺失。在这种情况下,MRM和PRM最有效地确定了对抑制剂治疗的整体运动学反应,突显了先验靶点鉴定和定量蛋白质组学数据集手动评估的价值。我们比较了从PRM,MRM,










































 京公网安备 11010802027423号
京公网安备 11010802027423号