当前位置:
X-MOL 学术
›
Mol. Syst. Des. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Accurate density functional theory (DFT) protocol for screening and designing chain transfer and branching agents for LDPE systems†
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2017-11-21 00:00:00 , DOI: 10.1039/c7me00087a Ivan Konstantinov 1, 2, 3 , Sean Ewart 1, 2, 3 , Hayley Brown 1, 2, 3 , Christopher Eddy 1, 2, 3 , Jonathan Mendenhall 1, 2, 3 , Sarat Munjal 1, 2, 3
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2017-11-21 00:00:00 , DOI: 10.1039/c7me00087a Ivan Konstantinov 1, 2, 3 , Sean Ewart 1, 2, 3 , Hayley Brown 1, 2, 3 , Christopher Eddy 1, 2, 3 , Jonathan Mendenhall 1, 2, 3 , Sarat Munjal 1, 2, 3
Affiliation
|
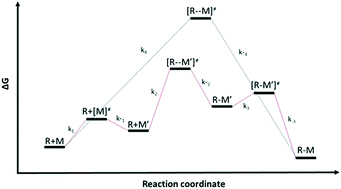
In this work, a density functional theory (DFT) methodology was developed and validated against experimental data for relative hydrogen abstraction (Cs) and monomer reactivity ratio (r1) parameters associated with free radical polymerization. For hydrogen abstraction, we considered ethane, cyclohexane, 2-butanone, propylene, isobutene, isobutane and propanal while methyl methacrylate, vinyl acetate, 1-butene, propylene and isobutene were the molecules of choice for benchmarking r1. It was shown that the M06-2X/6-311+G(3df,2p)//B3LYP/6-31+G(d,p) level of theory along with the counterpoise correction for the basis set superposition error (BSSE) produced estimated values in excellent agreement with experimental data. The calculated parameters were within a factor of 1.5 from the experimental values. This translated into a maximum error of 0.32 kcal mol−1 in Gibbs free energy of activation difference. The only exception was Cs for ethane with an experimental-to-calculated ratio of 3.0. Even then, the DFT estimate was within the experimental error. Furthermore, the approach managed to capture a wide range of empirical parameters as well as distinguish between monomers with close values. This robust and computationally inexpensive method can be applied to elucidate the reactivity of much larger species of industrial importance and rationally design the next generation of branching and chain-transfer agents for low density polyethylene (LDPE) systems.
中文翻译:

精确密度泛函理论(DFT)协议,用于筛选和设计LDPE系统的链转移和支化剂†
在这项工作中,开发了密度泛函理论(DFT)方法,并针对与自由基聚合相关的相对氢提取(C s)和单体反应率(r 1)参数的实验数据进行了验证。对于夺氢,我们考虑了乙烷,环己烷,2-丁酮,丙烯,异丁烯,异丁烷和丙醛,而甲基丙烯酸甲酯,乙酸乙烯酯,1-丁烯,丙烯和异丁烯是基准r 1的选择分子。。结果表明,M06-2X / 6-311 + G(3df,2p)// B3LYP / 6-31 + G(d,p)的理论水平以及对基组叠加误差(BSSE)的平衡校正产生的估计值与实验数据非常吻合。计算出的参数与实验值相差不到1.5倍。这转化为吉布斯活化能差的最大误差为0.32 kcal mol -1。唯一的例外是C s乙烷与实验计算值之比为3.0。即使这样,DFT估算值仍在实验误差范围内。此外,该方法设法捕获了广泛的经验参数,并区分了具有接近值的单体。这种健壮且计算便宜的方法可用于阐明具有重要工业意义的更大种类的反应性,并合理地设计用于低密度聚乙烯(LDPE)系统的下一代支化剂和链转移剂。
更新日期:2017-11-21
中文翻译:
精确密度泛函理论(DFT)协议,用于筛选和设计LDPE系统的链转移和支化剂†
在这项工作中,开发了密度泛函理论(DFT)方法,并针对与自由基聚合相关的相对氢提取(C s)和单体反应率(r 1)参数的实验数据进行了验证。对于夺氢,我们考虑了乙烷,环己烷,2-丁酮,丙烯,异丁烯,异丁烷和丙醛,而甲基丙烯酸甲酯,乙酸乙烯酯,1-丁烯,丙烯和异丁烯是基准r 1的选择分子。。结果表明,M06-2X / 6-311 + G(3df,2p)// B3LYP / 6-31 + G(d,p)的理论水平以及对基组叠加误差(BSSE)的平衡校正产生的估计值与实验数据非常吻合。计算出的参数与实验值相差不到1.5倍。这转化为吉布斯活化能差的最大误差为0.32 kcal mol -1。唯一的例外是C s乙烷与实验计算值之比为3.0。即使这样,DFT估算值仍在实验误差范围内。此外,该方法设法捕获了广泛的经验参数,并区分了具有接近值的单体。这种健壮且计算便宜的方法可用于阐明具有重要工业意义的更大种类的反应性,并合理地设计用于低密度聚乙烯(LDPE)系统的下一代支化剂和链转移剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号