当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Development of spin-orbit coupling for stochastic configuration interaction techniques
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2017-11-19 , DOI: 10.1002/jcc.25110 Paul Murphy 1 , Jeremy P. Coe 1 , Martin J. Paterson 1
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2017-11-19 , DOI: 10.1002/jcc.25110 Paul Murphy 1 , Jeremy P. Coe 1 , Martin J. Paterson 1
Affiliation
|
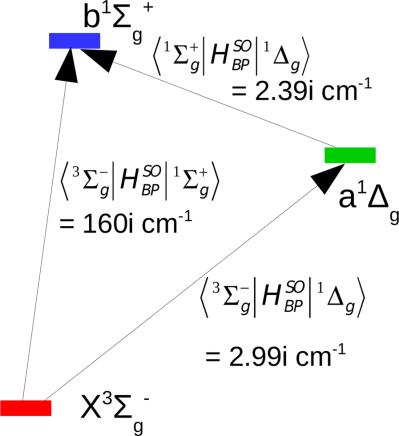
To perform spin‐orbit coupling calculations on atoms and molecules, good zeroth‐order wavefunctions are necessary. Here, we present the software development of the Monte Carlo Configuration Interaction (MCCI) method, to enable calculation of such properties, where MCCI iteratively constructs a multireference wavefunction using a stochastic procedure. In this initial work, we aim to establish the efficacy of this technique in predicting the splitting of otherwise degenerate energy levels on a range of atoms and small diatomic molecules. It is hoped that this work will subsequently act as a gateway toward using this method to investigate singlet‐triplet interactions in larger multireference molecules. We show that MCCI can generate very good results using highly compact wavefunctions compared to other techniques, with no prior knowledge of important orbitals. Higher‐order relativistic effects are neglected and spin‐orbit coupling effects are incorporated using first‐order degenerate perturbation theory with the Breit‐Pauli Hamiltonian and effective nuclear charges in the one‐electron operator. Results are obtained and presented for B, C, O, F, Si, S, and Cl atoms and OH, CN, NO, and C2 diatomic radicals including spin‐orbit coupling constants and the relative splitting of the lowest energy degenerate state for each species. Convergence of MCCI to the full configuration interaction result is demonstrated on the multireference problem of stretched OH. We also present results from the singlet‐triplet interaction between the X3Σg− and both the a1Δg and b1Σg+ states of the O2 molecule. © 2017 Wiley Periodicals, Inc.
中文翻译:

用于随机构型相互作用技术的自旋轨道耦合的发展
为了对原子和分子进行自旋轨道耦合计算,良好的零阶波函数是必要的。在这里,我们介绍了蒙特卡洛配置交互 (MCCI) 方法的软件开发,以实现此类属性的计算,其中 MCCI 使用随机程序迭代构建多参考波函数。在这项初步工作中,我们的目标是建立该技术在预测一系列原子和小双原子分子上其他简并能级分裂方面的功效。希望这项工作随后将成为使用这种方法研究更大的多参考分子中单线态-三线态相互作用的途径。我们表明,与其他技术相比,MCCI 可以使用高度紧凑的波函数产生非常好的结果,没有重要轨道的先验知识。高阶相对论效应被忽略,自旋轨道耦合效应被结合使用一阶简并微扰理论与 Breit-Pauli 哈密顿量和单电子算子中的有效核电荷。获得并展示了 B、C、O、F、Si、S 和 Cl 原子以及 OH、CN、NO 和 C2 双原子自由基的结果,包括自旋轨道耦合常数和最低能量简并态的相对分裂。物种。在拉伸 OH 的多参考问题上证明了 MCCI 对完整配置交互结果的收敛性。我们还展示了 X3Σg− 与 O2 分子的 a1Δg 和 b1Σg+ 状态之间的单线态-三重态相互作用的结果。© 2017 威利期刊公司。高阶相对论效应被忽略,自旋轨道耦合效应被结合使用一阶简并微扰理论与 Breit-Pauli 哈密顿量和单电子算子中的有效核电荷。获得并展示了 B、C、O、F、Si、S 和 Cl 原子以及 OH、CN、NO 和 C2 双原子自由基的结果,包括自旋轨道耦合常数和最低能量简并态的相对分裂。物种。在拉伸 OH 的多参考问题上证明了 MCCI 对完整配置交互结果的收敛性。我们还展示了 X3Σg− 与 O2 分子的 a1Δg 和 b1Σg+ 状态之间的单线态-三重态相互作用的结果。© 2017 威利期刊公司。高阶相对论效应被忽略,自旋轨道耦合效应被结合使用一阶简并微扰理论与 Breit-Pauli 哈密顿量和单电子算子中的有效核电荷。获得并展示了 B、C、O、F、Si、S 和 Cl 原子以及 OH、CN、NO 和 C2 双原子自由基的结果,包括自旋轨道耦合常数和最低能量简并态的相对分裂。物种。在拉伸 OH 的多参考问题上证明了 MCCI 对完整配置交互结果的收敛性。我们还展示了 X3Σg− 与 O2 分子的 a1Δg 和 b1Σg+ 状态之间的单线态-三重态相互作用的结果。© 2017 威利期刊公司。
更新日期:2017-11-19
中文翻译:
用于随机构型相互作用技术的自旋轨道耦合的发展
为了对原子和分子进行自旋轨道耦合计算,良好的零阶波函数是必要的。在这里,我们介绍了蒙特卡洛配置交互 (MCCI) 方法的软件开发,以实现此类属性的计算,其中 MCCI 使用随机程序迭代构建多参考波函数。在这项初步工作中,我们的目标是建立该技术在预测一系列原子和小双原子分子上其他简并能级分裂方面的功效。希望这项工作随后将成为使用这种方法研究更大的多参考分子中单线态-三线态相互作用的途径。我们表明,与其他技术相比,MCCI 可以使用高度紧凑的波函数产生非常好的结果,没有重要轨道的先验知识。高阶相对论效应被忽略,自旋轨道耦合效应被结合使用一阶简并微扰理论与 Breit-Pauli 哈密顿量和单电子算子中的有效核电荷。获得并展示了 B、C、O、F、Si、S 和 Cl 原子以及 OH、CN、NO 和 C2 双原子自由基的结果,包括自旋轨道耦合常数和最低能量简并态的相对分裂。物种。在拉伸 OH 的多参考问题上证明了 MCCI 对完整配置交互结果的收敛性。我们还展示了 X3Σg− 与 O2 分子的 a1Δg 和 b1Σg+ 状态之间的单线态-三重态相互作用的结果。© 2017 威利期刊公司。高阶相对论效应被忽略,自旋轨道耦合效应被结合使用一阶简并微扰理论与 Breit-Pauli 哈密顿量和单电子算子中的有效核电荷。获得并展示了 B、C、O、F、Si、S 和 Cl 原子以及 OH、CN、NO 和 C2 双原子自由基的结果,包括自旋轨道耦合常数和最低能量简并态的相对分裂。物种。在拉伸 OH 的多参考问题上证明了 MCCI 对完整配置交互结果的收敛性。我们还展示了 X3Σg− 与 O2 分子的 a1Δg 和 b1Σg+ 状态之间的单线态-三重态相互作用的结果。© 2017 威利期刊公司。高阶相对论效应被忽略,自旋轨道耦合效应被结合使用一阶简并微扰理论与 Breit-Pauli 哈密顿量和单电子算子中的有效核电荷。获得并展示了 B、C、O、F、Si、S 和 Cl 原子以及 OH、CN、NO 和 C2 双原子自由基的结果,包括自旋轨道耦合常数和最低能量简并态的相对分裂。物种。在拉伸 OH 的多参考问题上证明了 MCCI 对完整配置交互结果的收敛性。我们还展示了 X3Σg− 与 O2 分子的 a1Δg 和 b1Σg+ 状态之间的单线态-三重态相互作用的结果。© 2017 威利期刊公司。









































 京公网安备 11010802027423号
京公网安备 11010802027423号