当前位置:
X-MOL 学术
›
ACS Med. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Identification of an Indazole-Based Pharmacophore for the Inhibition of FGFR Kinases Using Fragment-Led de Novo Design
ACS Medicinal Chemistry Letters ( IF 3.5 ) Pub Date : 2017-11-17 00:00:00 , DOI: 10.1021/acsmedchemlett.7b00349 Lewis D. Turner 1 , Abbey J. Summers 1 , Laura O. Johnson 1 , Margaret A. Knowles , Colin W. G. Fishwick 1
ACS Medicinal Chemistry Letters ( IF 3.5 ) Pub Date : 2017-11-17 00:00:00 , DOI: 10.1021/acsmedchemlett.7b00349 Lewis D. Turner 1 , Abbey J. Summers 1 , Laura O. Johnson 1 , Margaret A. Knowles , Colin W. G. Fishwick 1
Affiliation
|
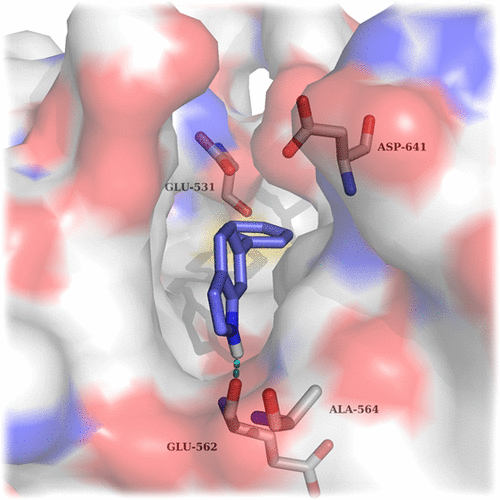
Structure-based drug design (SBDD) has become a powerful tool utilized by medicinal chemists to rationally guide the drug discovery process. Herein, we describe the use of SPROUT, a de novo-based program, to identify an indazole-based pharmacophore for the inhibition of fibroblast growth factor receptor (FGFR) kinases, which are validated targets for cancer therapy. Hit identification using SPROUT yielded 6-phenylindole as a small fragment predicted to bind to FGFR1. With the aid of docking models, several modifications to the indole were made to optimize the fragment to an indazole-containing pharmacophore, leading to a library of compounds containing 23 derivatives. Biological evaluation revealed that these indazole-containing fragments inhibited FGFR1–3 in the range of 0.8–90 μM with excellent ligand efficiencies of 0.30–0.48. Some compounds exhibited moderate selectivity toward individual FGFRs, indicating that further optimization using SBDD may lead to potent and selective inhibitors of the FGFR family.
中文翻译:

使用片段领导的de Novo设计鉴定基于吲唑的药基用于抑制FGFR激酶
基于结构的药物设计(SBDD)已成为药物化学家用来合理指导药物发现过程的强大工具。在此,我们描述从头开始使用SPROUT基于程序的程序,以识别基于吲唑的药效基团来抑制成纤维细胞生长因子受体(FGFR)激酶,这是经过验证的癌症治疗靶标。使用SPROUT进行命中鉴定可产生6-苯基吲哚,为预计会与FGFR1结合的小片段。借助于对接模型,对吲哚进行了几次修饰,以使片段最优化为含吲唑的药效团,从而形成了包含23种衍生物的化合物库。生物学评估表明,这些含吲唑的片段在0.8-90μM的范围内抑制FGFR1-3,具有出色的0.30-0.48配体效率。一些化合物对单个FGFR表现出中等选择性,这表明使用SBDD进行进一步优化可能会导致FGFR家族的有效和选择性抑制剂。
更新日期:2017-11-17
中文翻译:
使用片段领导的de Novo设计鉴定基于吲唑的药基用于抑制FGFR激酶
基于结构的药物设计(SBDD)已成为药物化学家用来合理指导药物发现过程的强大工具。在此,我们描述从头开始使用SPROUT基于程序的程序,以识别基于吲唑的药效基团来抑制成纤维细胞生长因子受体(FGFR)激酶,这是经过验证的癌症治疗靶标。使用SPROUT进行命中鉴定可产生6-苯基吲哚,为预计会与FGFR1结合的小片段。借助于对接模型,对吲哚进行了几次修饰,以使片段最优化为含吲唑的药效团,从而形成了包含23种衍生物的化合物库。生物学评估表明,这些含吲唑的片段在0.8-90μM的范围内抑制FGFR1-3,具有出色的0.30-0.48配体效率。一些化合物对单个FGFR表现出中等选择性,这表明使用SBDD进行进一步优化可能会导致FGFR家族的有效和选择性抑制剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号