当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Vanadium NMR Chemical Shifts of (Imido)vanadium(V) Dichloride Complexes with Imidazolin-2-iminato and Imidazolidin-2-iminato Ligands: Cooperation with Quantum-Chemical Calculations and Multiple Linear Regression Analyses
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-11-17 00:00:00 , DOI: 10.1021/acs.jpca.7b08328 Jun Yi 1 , Wenhong Yang 2 , Wen-Hua Sun 2 , Kotohiro Nomura 1 , Masahiko Hada 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-11-17 00:00:00 , DOI: 10.1021/acs.jpca.7b08328 Jun Yi 1 , Wenhong Yang 2 , Wen-Hua Sun 2 , Kotohiro Nomura 1 , Masahiko Hada 1
Affiliation
|
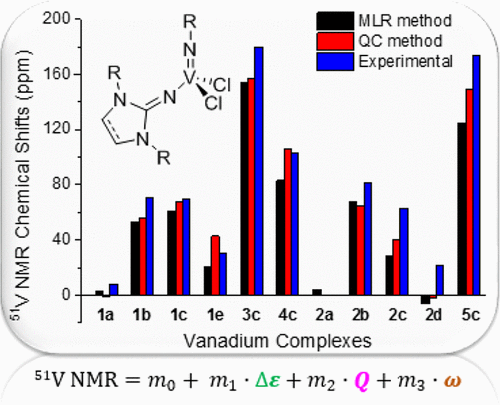
The NMR chemical shifts of vanadium (51V) in (imido)vanadium(V) dichloride complexes with imidazolin-2-iminato and imidazolidin-2-iminato ligands were calculated by the density functional theory (DFT) method with GIAO. The calculated 51V NMR chemical shifts were analyzed by the multiple linear regression (MLR) analysis (MLRA) method with a series of calculated molecular properties. Some of calculated NMR chemical shifts were incorrect using the optimized molecular geometries of the X-ray structures. After the global minimum geometries of all of the molecules were determined, the trend of the observed chemical shifts was well reproduced by the present DFT method. The MLRA method was performed to investigate the correlation between the 51V NMR chemical shift and the natural charge, band energy gap, and Wiberg bond index of the V═N bond. The 51V NMR chemical shifts obtained with the present MLR model were well reproduced with a correlation coefficient of 0.97.
中文翻译:

(Imido)钒(V)二氯化物与咪唑啉-2-亚氨基和咪唑啉二-2-亚氨基配体的钒NMR化学位移:与量子化学计算和多元线性回归分析的合作
通过GIAO的密度泛函理论(DFT)方法计算了在具有咪唑啉-2-亚氨基和咪唑啉二-2-亚氨基配体的(亚氨基)二氯化钒(V)络合物中钒(51 V)的NMR化学位移。通过多元线性回归(MLR)分析(MLRA)方法对计算出的51 V NMR化学位移进行分析,并计算出一系列分子特性。使用X射线结构的优化分子几何结构,某些计算出的NMR化学位移是不正确的。确定所有分子的整体最小几何形状后,通过本DFT方法可以很好地再现观察到的化学位移的趋势。进行了MLRA方法以研究51V NMR化学位移和V═N键的自然电荷,能带隙和Wiberg键指数。用本MLR模型获得的51 V NMR化学位移可以很好地再现,相关系数为0.97。
更新日期:2017-11-19
中文翻译:
(Imido)钒(V)二氯化物与咪唑啉-2-亚氨基和咪唑啉二-2-亚氨基配体的钒NMR化学位移:与量子化学计算和多元线性回归分析的合作
通过GIAO的密度泛函理论(DFT)方法计算了在具有咪唑啉-2-亚氨基和咪唑啉二-2-亚氨基配体的(亚氨基)二氯化钒(V)络合物中钒(51 V)的NMR化学位移。通过多元线性回归(MLR)分析(MLRA)方法对计算出的51 V NMR化学位移进行分析,并计算出一系列分子特性。使用X射线结构的优化分子几何结构,某些计算出的NMR化学位移是不正确的。确定所有分子的整体最小几何形状后,通过本DFT方法可以很好地再现观察到的化学位移的趋势。进行了MLRA方法以研究51V NMR化学位移和V═N键的自然电荷,能带隙和Wiberg键指数。用本MLR模型获得的51 V NMR化学位移可以很好地再现,相关系数为0.97。








































 京公网安备 11010802027423号
京公网安备 11010802027423号