当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Binding Thermodynamics and Kinetics Calculations Using Chemical Host and Guest: A Comprehensive Picture of Molecular Recognition
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-14 00:00:00 , DOI: 10.1021/acs.jctc.7b00899 Zhiye Tang 1 , Chia-En A Chang 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2017-12-14 00:00:00 , DOI: 10.1021/acs.jctc.7b00899 Zhiye Tang 1 , Chia-En A Chang 1
Affiliation
|
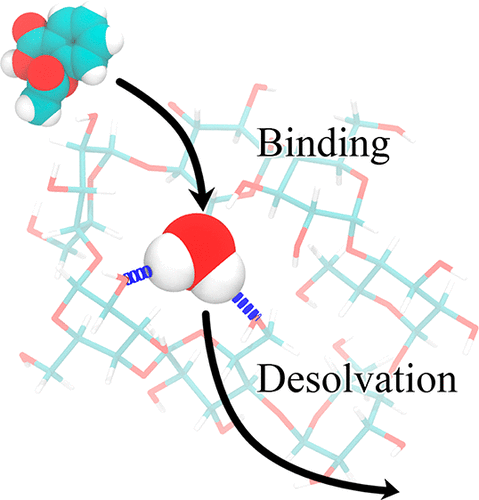
Understanding the fine balance between changes of entropy and enthalpy and the competition between a guest and water molecules in molecular binding is crucial in fundamental studies and practical applications. Experiments provide measurements. However, illustrating the binding/unbinding processes gives a complete picture of molecular recognition not directly available from experiments, and computational methods bridge the gaps. Here, we investigated guest association/dissociation with β-cyclodextrin (β-CD) by using microsecond-time-scale molecular dynamics (MD) simulations, postanalysis and numerical calculations. We computed association and dissociation rate constants, enthalpy, and solvent and solute entropy of binding. All the computed values of kon, koff, ΔH, ΔS, and ΔG using GAFF-CD and q4MD-CD force fields for β-CD could be compared with experimental data directly and agreed reasonably with experiment findings. In addition, our study further interprets experiments. Both force fields resulted in similar computed ΔG from independently computed kinetics rates, ΔG = −RT ln(kon·C0/koff), and thermodynamics properties, ΔG = ΔH – TΔS. The water entropy calculations show that the entropy gain of desolvating water molecules are a major driving force, and both force fields have the same strength of nonpolar attractions between solutes and β-CD as well. Water molecules play a crucial role in guest binding to β-CD. However, collective water/β-CD motions could contribute to different computed kon and ΔH values by different force fields, mainly because the parameters of β-CD provide different motions of β-CD, hydrogen-bond networks of water molecules in the cavity of free β-CD, and strength of desolvation penalty. As a result, q4MD-CD suggests that guest binding is mostly driven by enthalpy, while GAFF-CD shows that gaining entropy is the major driving force of binding. The study deepens our understanding of ligand–receptor recognition and suggests strategies for force field parametrization for accurately modeling molecular systems.
中文翻译:

使用化学主体和客体进行结合热力学和动力学计算:分子识别的全面图景
了解熵和焓的变化以及分子结合中客体和水分子之间的竞争之间的良好平衡对于基础研究和实际应用至关重要。实验提供测量结果。然而,说明结合/解除结合过程给出了分子识别的完整图像,而这些分子识别不能直接从实验中获得,而计算方法弥补了差距。在这里,我们通过使用微秒级分子动力学 (MD) 模拟、后分析和数值计算研究了客体与 β-环糊精 (β-CD) 的缔合/解离。我们计算了缔合和解离速率常数、焓以及溶剂和溶质的结合熵。使用 GAFF-CD 和 q4MD-CD 力场计算 β-CD 的k on 、 k off 、Δ H 、Δ S和 Δ G的所有计算值都可以直接与实验数据进行比较,并与实验结果合理吻合。此外,我们的研究进一步解释了实验。两个力场根据独立计算的动力学速率 Δ G = − RT ln( k on · C 0 / k off ) 和热力学性质 Δ G = Δ H – T Δ S得出相似的计算 Δ G 。水熵计算表明,去溶剂化水分子的熵增益是主要驱动力,并且两个力场在溶质和β-CD之间具有相同强度的非极性吸引力。水分子在客体与 β-CD 的结合中起着至关重要的作用。 然而,水/β-CD 的集体运动可能会通过不同的力场导致不同的计算k on和 Δ H值,这主要是因为 β-CD 的参数提供了 β-CD 的不同运动,水分子的氢键网络游离 β-CD 的空腔和去溶剂化惩罚的强度。因此,q4MD-CD表明客体结合主要由焓驱动,而GAFF-CD表明获得熵是结合的主要驱动力。该研究加深了我们对配体-受体识别的理解,并提出了用于精确建模分子系统的力场参数化策略。
更新日期:2017-12-14
中文翻译:
使用化学主体和客体进行结合热力学和动力学计算:分子识别的全面图景
了解熵和焓的变化以及分子结合中客体和水分子之间的竞争之间的良好平衡对于基础研究和实际应用至关重要。实验提供测量结果。然而,说明结合/解除结合过程给出了分子识别的完整图像,而这些分子识别不能直接从实验中获得,而计算方法弥补了差距。在这里,我们通过使用微秒级分子动力学 (MD) 模拟、后分析和数值计算研究了客体与 β-环糊精 (β-CD) 的缔合/解离。我们计算了缔合和解离速率常数、焓以及溶剂和溶质的结合熵。使用 GAFF-CD 和 q4MD-CD 力场计算 β-CD 的k on 、 k off 、Δ H 、Δ S和 Δ G的所有计算值都可以直接与实验数据进行比较,并与实验结果合理吻合。此外,我们的研究进一步解释了实验。两个力场根据独立计算的动力学速率 Δ G = − RT ln( k on · C 0 / k off ) 和热力学性质 Δ G = Δ H – T Δ S得出相似的计算 Δ G 。水熵计算表明,去溶剂化水分子的熵增益是主要驱动力,并且两个力场在溶质和β-CD之间具有相同强度的非极性吸引力。水分子在客体与 β-CD 的结合中起着至关重要的作用。 然而,水/β-CD 的集体运动可能会通过不同的力场导致不同的计算k on和 Δ H值,这主要是因为 β-CD 的参数提供了 β-CD 的不同运动,水分子的氢键网络游离 β-CD 的空腔和去溶剂化惩罚的强度。因此,q4MD-CD表明客体结合主要由焓驱动,而GAFF-CD表明获得熵是结合的主要驱动力。该研究加深了我们对配体-受体识别的理解,并提出了用于精确建模分子系统的力场参数化策略。










































 京公网安备 11010802027423号
京公网安备 11010802027423号