当前位置:
X-MOL 学术
›
Biochemistry
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Combining H/D Exchange Mass Spectrometry and Computational Docking To Derive the Structure of Protein–Protein Complexes
Biochemistry ( IF 2.9 ) Pub Date : 2017-11-16 00:00:00 , DOI: 10.1021/acs.biochem.7b00643 Victoria A. Roberts 1 , Michael E. Pique 2 , Simon Hsu 3 , Sheng Li 3
Biochemistry ( IF 2.9 ) Pub Date : 2017-11-16 00:00:00 , DOI: 10.1021/acs.biochem.7b00643 Victoria A. Roberts 1 , Michael E. Pique 2 , Simon Hsu 3 , Sheng Li 3
Affiliation
|
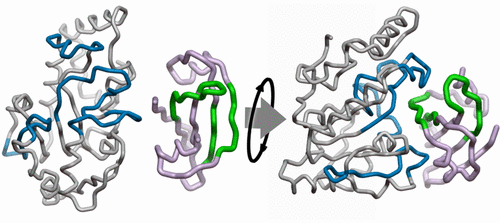
Protein–protein interactions are essential for biological function, but structures of protein–protein complexes are difficult to obtain experimentally. To derive the protein complex of the DNA-repair enzyme human uracil-DNA-glycosylase (hUNG) with its protein inhibitor (UGI), we combined rigid-body computational docking with hydrogen/deuterium exchange mass spectrometry (DXMS). Computational docking of the unbound protein structures provides a list of possible three-dimensional models of the complex; DXMS identifies solvent-protected protein residues. DXMS showed that unbound hUNG is compactly folded, but unbound UGI is loosely packed. An increased level of solvent protection of hUNG in the complex was localized to four regions on the same face. The decrease in the number of incorporated deuterons was quantitatively interpreted as the minimum number of main-chain hUNG amides buried in the protein–protein interface. The level of deuteration of complexed UGI decreased throughout the protein chain, indicating both tighter packing and direct solvent protection by hUNG. Three UGI regions showing the greatest decreases were best interpreted leniently, requiring just one main-chain amide from each in the interface. Applying the DXMS constraints as filters to a list of docked complexes gave the correct complex as the largest favorable energy cluster. Thus, identification of approximate protein interfaces was sufficient to distinguish the protein complex. Surprisingly, incorporating the DXMS data as added favorable potentials in the docking calculation was less effective in finding the correct complex. The filtering method has greater flexibility, with the capability to test each constraint and enforce simultaneous contact by multiple regions, but with the caveat that the list from the unbiased docking must include correct complexes.
中文翻译:

结合H / D交换质谱和计算对接得出蛋白质-蛋白质复合物的结构
蛋白质-蛋白质相互作用对于生物学功能至关重要,但是蛋白质-蛋白质复合物的结构很难通过实验获得。为了获得具有蛋白质抑制剂(UGI)的DNA修复酶人尿嘧啶DNA糖基化酶(hUNG)的蛋白质复合物,我们将刚体计算对接与氢/氘交换质谱(DXMS)结合在一起。未结合蛋白结构的计算对接提供了复合物可能的三维模型的列表;DXMS识别溶剂保护的蛋白质残基。DXMS显示未结合的hUNG紧密折叠,但未结合的UGI松散包装。在复合物中,hUNG溶剂保护水平的提高被定位在同一面上的四个区域。掺入氘核数目的减少被定量解释为埋在蛋白质-蛋白质界面中的主链hUNG酰胺的最小数目。复杂的UGI的氘化水平在整个蛋白质链中均降低,表明紧密包装和hUNG的直接溶剂保护作用。宽大地解释了三个显示最大减少的UGI区域,只需在界面中每个区域仅需要一个主链酰胺。将DXMS约束作为过滤器应用到对接的配合物列表中,可以将正确的配合物作为最大的有利能量簇。因此,鉴定近似的蛋白质界面足以区分蛋白质复合物。出奇,将DXMS数据作为对接计算中增加的有利电位在寻找正确的复合物时不太有效。筛选方法具有更大的灵活性,能够测试每个约束并强制多个区域同时进行接触,但需要注意的是,无偏对接中的列表必须包含正确的复合体。
更新日期:2017-11-17
中文翻译:
结合H / D交换质谱和计算对接得出蛋白质-蛋白质复合物的结构
蛋白质-蛋白质相互作用对于生物学功能至关重要,但是蛋白质-蛋白质复合物的结构很难通过实验获得。为了获得具有蛋白质抑制剂(UGI)的DNA修复酶人尿嘧啶DNA糖基化酶(hUNG)的蛋白质复合物,我们将刚体计算对接与氢/氘交换质谱(DXMS)结合在一起。未结合蛋白结构的计算对接提供了复合物可能的三维模型的列表;DXMS识别溶剂保护的蛋白质残基。DXMS显示未结合的hUNG紧密折叠,但未结合的UGI松散包装。在复合物中,hUNG溶剂保护水平的提高被定位在同一面上的四个区域。掺入氘核数目的减少被定量解释为埋在蛋白质-蛋白质界面中的主链hUNG酰胺的最小数目。复杂的UGI的氘化水平在整个蛋白质链中均降低,表明紧密包装和hUNG的直接溶剂保护作用。宽大地解释了三个显示最大减少的UGI区域,只需在界面中每个区域仅需要一个主链酰胺。将DXMS约束作为过滤器应用到对接的配合物列表中,可以将正确的配合物作为最大的有利能量簇。因此,鉴定近似的蛋白质界面足以区分蛋白质复合物。出奇,将DXMS数据作为对接计算中增加的有利电位在寻找正确的复合物时不太有效。筛选方法具有更大的灵活性,能够测试每个约束并强制多个区域同时进行接触,但需要注意的是,无偏对接中的列表必须包含正确的复合体。








































 京公网安备 11010802027423号
京公网安备 11010802027423号