当前位置:
X-MOL 学术
›
CrystEngComm
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The impact of vinylene bridges and side chain alkyl groups on the solid state structures of tricyanovinyl-substituted thiophenes†
CrystEngComm ( IF 2.6 ) Pub Date : 2017-11-16 00:00:00 , DOI: 10.1039/c7ce01574g Phuong-Truc T. Pham 1, 2, 3 , Victor G. Young 1, 3, 4, 5 , Mamoun M. Bader 1, 6, 7, 8
CrystEngComm ( IF 2.6 ) Pub Date : 2017-11-16 00:00:00 , DOI: 10.1039/c7ce01574g Phuong-Truc T. Pham 1, 2, 3 , Victor G. Young 1, 3, 4, 5 , Mamoun M. Bader 1, 6, 7, 8
Affiliation
|
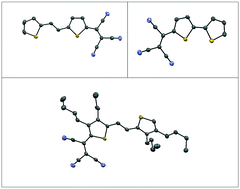
The goal of this work is to examine the solid state structures of compounds that have been designed for increased conjugation and solubility, as these factors are important if these compounds are to be used in the solid state. The impact of three commonly employed molecular design strategies on the solid state structures of three thiophene derivatives is reported herein. These strategies include: (i) introduction of a strong electron accepting group (2T–TCV, 1); (ii) increase in conjugation by introducing a vinylene bridge in the presence of a strong electron accepting group (2T–TCV with both a TCV group and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bridge, 2); and (iii) enhancing the solubility by introducing n-butyl side chain groups in the presence of both a strong electron accepting group and a CC bridge (2T–TCV containing a strong electron accepting group, a CC bridge and four n-butyl groups, 3). Compounds 1 and 2 crystallize with four molecules in the unit cell while the unit cell of compound 3 contains only two molecules. The torsion between the two thiophene rings increases from 4.39° to 5.50° to 5.75° for 1, 2, and 3, respectively. The short distances between adjacent molecules within the unit cell also increase from 2.84 Å in 2 to 3.47 Å in 3. We also note that while the sulfur atoms assume a syn conformation in both 1 and 2, they favor the anti-conformation in 3. DFT calculations show a small energy difference between the syn and anti-conformation for 1 and 2, i.e. 3.18 kJ mol−1 and 3.19 kJ mol−1, respectively; this energy difference is found to be greater for compound 3 with the anti-conformation being 17.47 kJ mol−1 more stable than the syn conformation.
C bridge, 2); and (iii) enhancing the solubility by introducing n-butyl side chain groups in the presence of both a strong electron accepting group and a CC bridge (2T–TCV containing a strong electron accepting group, a CC bridge and four n-butyl groups, 3). Compounds 1 and 2 crystallize with four molecules in the unit cell while the unit cell of compound 3 contains only two molecules. The torsion between the two thiophene rings increases from 4.39° to 5.50° to 5.75° for 1, 2, and 3, respectively. The short distances between adjacent molecules within the unit cell also increase from 2.84 Å in 2 to 3.47 Å in 3. We also note that while the sulfur atoms assume a syn conformation in both 1 and 2, they favor the anti-conformation in 3. DFT calculations show a small energy difference between the syn and anti-conformation for 1 and 2, i.e. 3.18 kJ mol−1 and 3.19 kJ mol−1, respectively; this energy difference is found to be greater for compound 3 with the anti-conformation being 17.47 kJ mol−1 more stable than the syn conformation.
中文翻译:

亚乙烯基桥和侧链烷基对三氰基乙烯基取代的噻吩固态结构的影响†
这项工作的目的是检查已设计用于增加缀合和溶解度的化合物的固态结构,因为如果这些化合物以固态使用,这些因素很重要。本文报道了三种常用的分子设计策略对三种噻吩衍生物的固态结构的影响。这些策略包括:(i)引入强电子接受基团(2T–TCV,1);(ii)通过在强电子接受基团(同时具有TCV基团和CC桥基的2T–TCV ,2)存在下引入亚乙烯基桥来增加共轭;(iii)通过引入n来提高溶解度-丁基侧链基团同时存在一个强电子接受基和一个C C桥(2T–TCV包含一个强电子接受基,一个C C桥和四个正丁基,3)。化合物1和2在晶胞中以四个分子结晶,而化合物3的晶胞仅包含两个分子。从4.39两个噻吩环的增加°〜5.50°〜5.75°,之间的扭转1,2,和3,分别。晶胞内相邻分子之间的短距离也从2中的2.84Å增加到3中的3.47Å。我们还注意到,尽管硫原子在1和2中均呈顺式构象,但它们在3中有利于反构象。DFT计算表明,对于1和2,顺构和反构象之间的能量差较小,即分别为3.18 kJ mol -1和3.19 kJ mol -1。发现化合物3的该能量差更大,反构象比顺式构象更稳定,反构象为17.47 kJ mol -1。
更新日期:2017-11-16
C bridge, 2); and (iii) enhancing the solubility by introducing n-butyl side chain groups in the presence of both a strong electron accepting group and a CC bridge (2T–TCV containing a strong electron accepting group, a CC bridge and four n-butyl groups, 3). Compounds 1 and 2 crystallize with four molecules in the unit cell while the unit cell of compound 3 contains only two molecules. The torsion between the two thiophene rings increases from 4.39° to 5.50° to 5.75° for 1, 2, and 3, respectively. The short distances between adjacent molecules within the unit cell also increase from 2.84 Å in 2 to 3.47 Å in 3. We also note that while the sulfur atoms assume a syn conformation in both 1 and 2, they favor the anti-conformation in 3. DFT calculations show a small energy difference between the syn and anti-conformation for 1 and 2, i.e. 3.18 kJ mol−1 and 3.19 kJ mol−1, respectively; this energy difference is found to be greater for compound 3 with the anti-conformation being 17.47 kJ mol−1 more stable than the syn conformation.
中文翻译:
亚乙烯基桥和侧链烷基对三氰基乙烯基取代的噻吩固态结构的影响†
这项工作的目的是检查已设计用于增加缀合和溶解度的化合物的固态结构,因为如果这些化合物以固态使用,这些因素很重要。本文报道了三种常用的分子设计策略对三种噻吩衍生物的固态结构的影响。这些策略包括:(i)引入强电子接受基团(2T–TCV,1);(ii)通过在强电子接受基团(同时具有TCV基团和C
C桥基的2T–TCV ,2)存在下引入亚乙烯基桥来增加共轭;(iii)通过引入n来提高溶解度-丁基侧链基团同时存在一个强电子接受基和一个C C桥(2T–TCV包含一个强电子接受基,一个C C桥和四个正丁基,3)。化合物1和2在晶胞中以四个分子结晶,而化合物3的晶胞仅包含两个分子。从4.39两个噻吩环的增加°〜5.50°〜5.75°,之间的扭转1,2,和3,分别。晶胞内相邻分子之间的短距离也从2中的2.84Å增加到3中的3.47Å。我们还注意到,尽管硫原子在1和2中均呈顺式构象,但它们在3中有利于反构象。DFT计算表明,对于1和2,顺构和反构象之间的能量差较小,即分别为3.18 kJ mol -1和3.19 kJ mol -1。发现化合物3的该能量差更大,反构象比顺式构象更稳定,反构象为17.47 kJ mol -1。










































 京公网安备 11010802027423号
京公网安备 11010802027423号