当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Methods for exploring reaction space in molecular systems
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2017-11-16 , DOI: 10.1002/wcms.1354 Amanda L. Dewyer 1 , Alonso J. Argüelles 1 , Paul M. Zimmerman 1
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2017-11-16 , DOI: 10.1002/wcms.1354 Amanda L. Dewyer 1 , Alonso J. Argüelles 1 , Paul M. Zimmerman 1
Affiliation
|
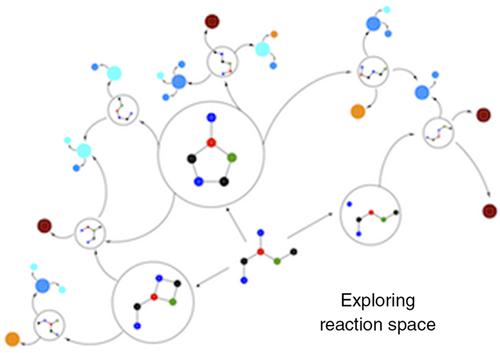
The area of reaction mechanism discovery simulation has taken considerable strides in recent years. Novel methods that make hypotheses for elementary steps and complementary means for reaction path and transition state (TS) optimization are lowering the amount of chemical intuition and user effort required to explore reaction networks. The resulting networks lead from reactants to reactive intermediates and products, and are becoming closer representations of physical mechanisms involved in experiments. This review describes several of these approaches, which are categorized based on their overarching TS finding strategies. Future advances are discussed that may revolutionize the ability of simulation to fully predict not just the reaction mechanism but reaction outcomes. WIREs Comput Mol Sci 2018, 8:e1354. doi: 10.1002/wcms.1354
中文翻译:

探索分子系统反应空间的方法
近年来,反应机理发现模拟领域取得了长足的进步。提出基本步骤假设的新方法以及反应路径和过渡态(TS)优化的补充手段正在降低化学直觉的数量和探索反应网络所需的用户工作量。生成的网络从反应物引向反应性中间体和产物,并且正在越来越接近地代表实验所涉及的物理机制。这篇综述描述了其中的几种方法,这些方法根据其总体TS查找策略进行了分类。讨论了未来的进展,这些进展可能会彻底改变模拟功能,使其不仅可以完全预测反应机理,而且可以完全预测反应结果。电线计算机科学2018,8:e1354。doi:10.1002 / wcms.1354
更新日期:2017-11-16
中文翻译:
探索分子系统反应空间的方法
近年来,反应机理发现模拟领域取得了长足的进步。提出基本步骤假设的新方法以及反应路径和过渡态(TS)优化的补充手段正在降低化学直觉的数量和探索反应网络所需的用户工作量。生成的网络从反应物引向反应性中间体和产物,并且正在越来越接近地代表实验所涉及的物理机制。这篇综述描述了其中的几种方法,这些方法根据其总体TS查找策略进行了分类。讨论了未来的进展,这些进展可能会彻底改变模拟功能,使其不仅可以完全预测反应机理,而且可以完全预测反应结果。电线计算机科学2018,8:e1354。doi:10.1002 / wcms.1354










































 京公网安备 11010802027423号
京公网安备 11010802027423号