当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Enhancing Intramolecular Chalcogen Interactions in 1-Hydroxy-8-YH-naphthalene Derivatives
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-11-10 00:00:00 , DOI: 10.1021/acs.jpca.7b09678 Goar Sánchez-Sanz 1 , Cristina Trujillo 2 , Ibon Alkorta 3 , José Elguero 3
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-11-10 00:00:00 , DOI: 10.1021/acs.jpca.7b09678 Goar Sánchez-Sanz 1 , Cristina Trujillo 2 , Ibon Alkorta 3 , José Elguero 3
Affiliation
|
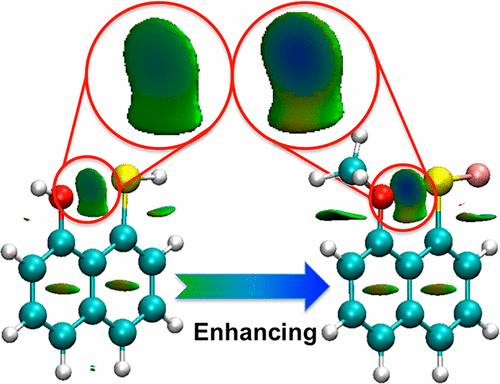
Forty-two peri-substituted naphthalene derivatives presenting chalcogen weak interactions were studied. They correspond to O···Y interactions, Y being O, S, and Se. While the O atom bears H or CH3 substituents (OH and OCH3 groups), the Y atom is substituted by H, F, and CN to explore the effect of these electron-donating and electron-withdrawing substituents on the chalcogen bond strength. The effect of F and CH3 substituents on positions ortho/para (2,4,5,7 of the naphthalene ring) was also studied. Optimizations were performed at the MP2/aug-cc-pVDZ, and binding energies were performed at the MP2/aug-cc-pVDZ followed by an MP2/CBS estimation. The main properties studied were geometries, energies (Eb, Eiso, and Edef), the molecular electrostatic potential, electron density shifts, natural bond order E(2) energies, and the relationship between these properties.
中文翻译:

增强1-Hydroxy-8-YH-萘衍生物中的分子内硫属元素相互作用
研究了42种被硫族元素弱相互作用的邻取代萘衍生物。它们对应于O···Y相互作用,Y为O,S和Se。当O原子带有H或CH 3取代基(OH和OCH 3基团)时,Y原子被H,F和CN取代,以探索这些给电子和吸电子取代基对硫族元素键强度的影响。还研究了F和CH 3取代基对邻位/对位(萘环的2,4,5,7)的影响。优化在MP2 / aug-cc-pVDZ处进行,结合能在MP2 / aug-cc-pVDZ处进行,然后进行MP2 / CBS估计。研究的主要特性是几何形状,能量(E b,Eiso和E def),分子静电势,电子密度移动,自然键序E(2)能量以及这些属性之间的关系。
更新日期:2017-11-11
中文翻译:
增强1-Hydroxy-8-YH-萘衍生物中的分子内硫属元素相互作用
研究了42种被硫族元素弱相互作用的邻取代萘衍生物。它们对应于O···Y相互作用,Y为O,S和Se。当O原子带有H或CH 3取代基(OH和OCH 3基团)时,Y原子被H,F和CN取代,以探索这些给电子和吸电子取代基对硫族元素键强度的影响。还研究了F和CH 3取代基对邻位/对位(萘环的2,4,5,7)的影响。优化在MP2 / aug-cc-pVDZ处进行,结合能在MP2 / aug-cc-pVDZ处进行,然后进行MP2 / CBS估计。研究的主要特性是几何形状,能量(E b,Eiso和E def),分子静电势,电子密度移动,自然键序E(2)能量以及这些属性之间的关系。










































 京公网安备 11010802027423号
京公网安备 11010802027423号