当前位置:
X-MOL 学术
›
J. Comput. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Hyperconjugative effects in π-hydrogen bonding: Theory and experiment
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2017-11-08 , DOI: 10.1002/jcc.25088 Boris Galabov 1, 2 , Valia Nikolova 1 , Diana Cheshmedzhieva 1 , Boriana Hadjieva 1 , Henry F. Schaefer 2
Journal of Computational Chemistry ( IF 3.4 ) Pub Date : 2017-11-08 , DOI: 10.1002/jcc.25088 Boris Galabov 1, 2 , Valia Nikolova 1 , Diana Cheshmedzhieva 1 , Boriana Hadjieva 1 , Henry F. Schaefer 2
Affiliation
|
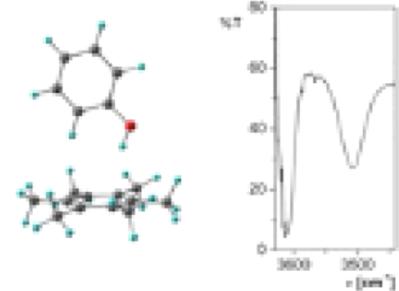
Density functional theory computations with the B3LYP/6‐311++G(2df,2p) method and IR spectroscopy are employed in investigating the properties of twenty π‐hydrogen bonded complexes between substituted phenols and hexamethylbenzene. All complexes possess T‐shaped structures. The methyl hyperconjugative effects on interactions energies and OH stretching frequencies are estimated via comparisons with previously reported theoretical and experimental results for analogous phenol complexes with benzene. The theoretical computations provide excellent quantitative predictions of the OH stretching frequency shifts (ΔνOH) resulting from the hydrogen bonding. The ΔνOH shifts in the hexamethylbenzene complexes are approximately twice as large as the corresponding shifts for the benzene complexes. Hirshfeld charges, electrostatic potential at nuclei values, and molecular electrostatic potential maps are employed in gaining insights into the mechanisms of methyl hyperconjugative effects on complex formation. © 2017 Wiley Periodicals, Inc.
中文翻译:

π-氢键中的超共轭效应:理论与实验
使用 B3LYP/6-311++G(2df,2p) 方法和红外光谱进行密度泛函理论计算,研究了取代苯酚和六甲苯之间 20 个 π-氢键配合物的性质。所有配合物都具有 T 形结构。甲基超共轭对相互作用能和 OH 伸缩频率的影响是通过与先前报道的类似苯酚配合物的理论和实验结果进行比较来估计的。理论计算提供了由氢键引起的 OH 伸缩频移 (ΔνOH) 的出色定量预测。六甲基苯配合物中的 ΔνOH 位移大约是苯配合物相应位移的两倍。Hirshfeld 电荷,核值处的静电势,分子静电势图用于深入了解甲基超共轭效应对复合物形成的机制。© 2017 威利期刊公司。
更新日期:2017-11-08
中文翻译:
π-氢键中的超共轭效应:理论与实验
使用 B3LYP/6-311++G(2df,2p) 方法和红外光谱进行密度泛函理论计算,研究了取代苯酚和六甲苯之间 20 个 π-氢键配合物的性质。所有配合物都具有 T 形结构。甲基超共轭对相互作用能和 OH 伸缩频率的影响是通过与先前报道的类似苯酚配合物的理论和实验结果进行比较来估计的。理论计算提供了由氢键引起的 OH 伸缩频移 (ΔνOH) 的出色定量预测。六甲基苯配合物中的 ΔνOH 位移大约是苯配合物相应位移的两倍。Hirshfeld 电荷,核值处的静电势,分子静电势图用于深入了解甲基超共轭效应对复合物形成的机制。© 2017 威利期刊公司。










































 京公网安备 11010802027423号
京公网安备 11010802027423号