当前位置:
X-MOL 学术
›
J. Chem. Inf. Model.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Bioisostere Identification by Determining the Amino Acid Binding Preferences of Common Chemical Fragments
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2017-12-01 00:00:00 , DOI: 10.1021/acs.jcim.7b00092 Tomohiro Sato 1 , Noriaki Hashimoto 2 , Teruki Honma 1
Journal of Chemical Information and Modeling ( IF 5.6 ) Pub Date : 2017-12-01 00:00:00 , DOI: 10.1021/acs.jcim.7b00092 Tomohiro Sato 1 , Noriaki Hashimoto 2 , Teruki Honma 1
Affiliation
|
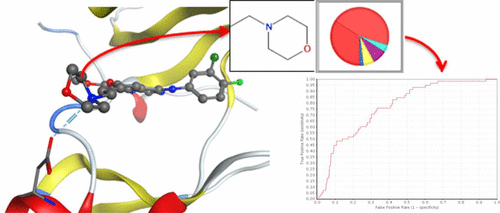
To assist in the structural optimization of hit/lead compounds during drug discovery, various computational approaches to identify potentially useful bioisosteric conversions have been reported. Here, the preference of chemical fragments to hydrogen bonds with specific amino acid residues was used to identify potential bioisosteric conversions. We first compiled a data set of chemical fragments frequently occurring in complex structures contained in the Protein Data Bank. We then used a computational approach to determine the amino acids to which these chemical fragments most frequently hydrogen bonded. The results of the frequency analysis were used to hierarchically cluster chemical fragments according to their amino acid preferences. The Euclid distance between amino acid preferences of chemical fragments for hydrogen bonding was then compared to MMP information in the ChEMBL database. To demonstrate the applicability of the approach for compound optimization, the similarity of amino acid preferences was used to identify known bioisosteric conversions of the epidermal growth factor receptor inhibitor gefitinib. The amino acid preference distance successfully detected bioisosteric fragments corresponding to the morpholine ring in gefitinib with a higher ROC score compared to those based on topological similarity of substituents and frequency of MMP in the ChEMBL database.
中文翻译:

通过确定常见化学片段的氨基酸结合偏好来确定生物等排体
为了在药物发现过程中协助命中/先导化合物的结构优化,已报道了各种计算方法来鉴定潜在有用的生物立体转换。在这里,化学片段对具有特定氨基酸残基的氢键的偏爱被用来确定潜在的生物立体转换。我们首先汇编了蛋白质片段数据库中包含的复杂结构中经常发生的化学片段数据集。然后,我们使用一种计算方法来确定这些化学片段最常与氢键合的氨基酸。频率分析的结果用于根据化学片段的氨基酸偏好对它们进行分层聚类。然后,在ChEMBL数据库中将化学片段的氢键键合氨基酸偏好之间的Euclid距离与MMP信息进行比较。为了证明该方法对化合物优化的适用性,使用氨基酸偏好的相似性来确定表皮生长因子受体抑制剂吉非替尼的已知生物等位转换。与基于ChEMBL数据库中取代基的拓扑相似性和MMP的频率相比,氨基酸优先距离成功地检测出了吉非替尼中对应于吗啉环的生物立体片段,其ROC得分更高。氨基酸偏好的相似性用于鉴定表皮生长因子受体抑制剂吉非替尼的已知生物立体转换。与基于ChEMBL数据库中取代基的拓扑相似性和MMP的频率相比,氨基酸优先距离成功地检测出了吉非替尼中对应于吗啉环的生物立体片段,其ROC得分更高。氨基酸偏好的相似性用于鉴定表皮生长因子受体抑制剂吉非替尼的已知生物立体转换。与基于ChEMBL数据库中取代基的拓扑相似性和MMP的频率相比,氨基酸优先距离成功地检测出了吉非替尼中对应于吗啉环的生物立体片段,其ROC得分更高。
更新日期:2017-12-01
中文翻译:
通过确定常见化学片段的氨基酸结合偏好来确定生物等排体
为了在药物发现过程中协助命中/先导化合物的结构优化,已报道了各种计算方法来鉴定潜在有用的生物立体转换。在这里,化学片段对具有特定氨基酸残基的氢键的偏爱被用来确定潜在的生物立体转换。我们首先汇编了蛋白质片段数据库中包含的复杂结构中经常发生的化学片段数据集。然后,我们使用一种计算方法来确定这些化学片段最常与氢键合的氨基酸。频率分析的结果用于根据化学片段的氨基酸偏好对它们进行分层聚类。然后,在ChEMBL数据库中将化学片段的氢键键合氨基酸偏好之间的Euclid距离与MMP信息进行比较。为了证明该方法对化合物优化的适用性,使用氨基酸偏好的相似性来确定表皮生长因子受体抑制剂吉非替尼的已知生物等位转换。与基于ChEMBL数据库中取代基的拓扑相似性和MMP的频率相比,氨基酸优先距离成功地检测出了吉非替尼中对应于吗啉环的生物立体片段,其ROC得分更高。氨基酸偏好的相似性用于鉴定表皮生长因子受体抑制剂吉非替尼的已知生物立体转换。与基于ChEMBL数据库中取代基的拓扑相似性和MMP的频率相比,氨基酸优先距离成功地检测出了吉非替尼中对应于吗啉环的生物立体片段,其ROC得分更高。氨基酸偏好的相似性用于鉴定表皮生长因子受体抑制剂吉非替尼的已知生物立体转换。与基于ChEMBL数据库中取代基的拓扑相似性和MMP的频率相比,氨基酸优先距离成功地检测出了吉非替尼中对应于吗啉环的生物立体片段,其ROC得分更高。


























 京公网安备 11010802027423号
京公网安备 11010802027423号