当前位置:
X-MOL 学术
›
Mol. Syst. Des. Eng.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Unfolding dynamics of small peptides biased by constant mechanical forces
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2017-10-30 00:00:00 , DOI: 10.1039/c7me00080d Fabian Knoch 1, 2, 3, 4 , Thomas Speck 1, 2, 3, 4
Molecular Systems Design & Engineering ( IF 3.2 ) Pub Date : 2017-10-30 00:00:00 , DOI: 10.1039/c7me00080d Fabian Knoch 1, 2, 3, 4 , Thomas Speck 1, 2, 3, 4
Affiliation
|
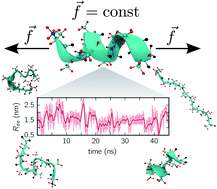
We show how multi-ensemble Markov state models can be combined with constant-force equilibrium simulations. Besides obtaining the unfolding/folding rates, Markov state models allow gaining detailed insights into the folding dynamics and pathways through identifying folding intermediates and misfolded structures. For two specific peptides, we demonstrate that the end-to-end distance is an insufficient reaction coordinate. This problem is alleviated through constructing models with multiple collective variables, for which we employ the time-lagged independent component analysis requiring only minimal prior knowledge. Our results show that combining Markov state models with constant-force simulations is a promising strategy to bridge the gap between simulation and experiments even for medium-sized biomolecules.
中文翻译:

受恒定机械力偏置的小肽的展开动力学
我们展示了如何将多集合马尔可夫状态模型与恒力平衡仿真相结合。除了获得展开/折叠速率外,马尔可夫状态模型还可以通过识别折叠中间体和错折叠结构来获得有关折叠动力学和路径的详细见解。对于两个特定的肽,我们证明了端到端的距离是不足的反应坐标。通过构建具有多个集合变量的模型可以缓解此问题,为此,我们采用了时间滞后的独立分量分析,只需要最少的先验知识即可。我们的结果表明,将马尔可夫状态模型与恒力模拟相结合是弥合模拟与实验之间差距的一种有前途的策略,即使对于中等大小的生物分子也是如此。
更新日期:2017-10-30
中文翻译:
受恒定机械力偏置的小肽的展开动力学
我们展示了如何将多集合马尔可夫状态模型与恒力平衡仿真相结合。除了获得展开/折叠速率外,马尔可夫状态模型还可以通过识别折叠中间体和错折叠结构来获得有关折叠动力学和路径的详细见解。对于两个特定的肽,我们证明了端到端的距离是不足的反应坐标。通过构建具有多个集合变量的模型可以缓解此问题,为此,我们采用了时间滞后的独立分量分析,只需要最少的先验知识即可。我们的结果表明,将马尔可夫状态模型与恒力模拟相结合是弥合模拟与实验之间差距的一种有前途的策略,即使对于中等大小的生物分子也是如此。










































 京公网安备 11010802027423号
京公网安备 11010802027423号