Bioorganic & Medicinal Chemistry Letters ( IF 2.5 ) Pub Date : 2017-10-28 , DOI: 10.1016/j.bmcl.2017.10.041 Ting Chen , Venkataswamy Sorna , Susie Choi , Lee Call , Jared Bearss , Kent Carpenter , Steven L. Warner , Sunil Sharma , David J. Bearss , Hariprasad Vankayalapati
|
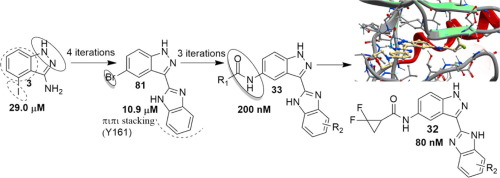
In this work, we describe the use of the rule of 3 fragment-based strategies from biochemical screening data of 1100 in-house, small, low molecular weight fragments. The sequential combination of in silico fragment hopping and fragment linking based on S160/Y161/A162 hinge residues hydrogen bonding interactions leads to the identification of novel 1H-benzo[d]imidazol-2-yl)-1H-indazol class of Phosphoinositide-Dependent Kinase-1 (PDK1) inhibitors. Consequent SAR and follow-up screening data led to the discovery of two potent PDK1 inhibitors: compound 32 and 35, with an IC50 of 80 nM and 94 nM, respectively. Further biological evaluation showed that, at the low nanomolar concentration, the drug had potent ability to inhibit phosphorylation of AKT and p70S6, and selectively kill the cancer cells with mutations in both PTEN and PI3K. The microarray data showed that DUSP6, DUSP4, and FOSL1 were down-regulated in the sensitive cell lines with the compound treatment. The in vivo test showed that 35 can significantly inhibit tumor growth without influencing body weight growth. Our results suggest that these compounds, especially 35, merit further pre-clinical evaluation.
中文翻译:
1 H-苯并[ d ]咪唑-2-基)-1 H-吲唑衍生物作为强效PDK1抑制剂的基于片段的设计,合成,生物学评估和比吸收比
在这项工作中,我们从1100个内部,小,低分子量片段的生化筛选数据中描述了3种基于片段策略的规则的使用。基于S160 / Y161 / A162铰链残基氢键相互作用的计算机片段跳跃和片段连接的顺序组合导致鉴定出新的1 H-苯并[ d ]咪唑-2-基)-1 H-吲唑类磷酸肌醇-依赖性激酶1(PDK1)抑制剂。随后的SAR和后续筛查数据导致发现了两种有效的PDK1抑制剂:化合物32和35,IC 50分别为80 nM和94 nM。进一步的生物学评估表明,在低纳摩尔浓度下,该药物具有强大的抑制AKT和p70S6磷酸化的能力,并选择性杀死具有PTEN和PI3K突变的癌细胞。微阵列数据显示,经过复合处理后,DUSP6,DUSP4和FOSL1在敏感细胞系中被下调。的体内试验表明,35可以显著抑制肿瘤的生长,而不影响体重的增长。我们的结果表明,这些化合物(尤其是35种)值得进一步的临床前评估。










































 京公网安备 11010802027423号
京公网安备 11010802027423号