当前位置:
X-MOL 学术
›
ChemMedChem
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Design, Synthesis, Molecular Modeling, and Biological Evaluation of Novel Amine‐based Histone Deacetylase Inhibitors
ChemMedChem ( IF 3.6 ) Pub Date : 2017-11-30 , DOI: 10.1002/cmdc.201700449 Hazem Abdelkarim 1 , Raghupathi Neelarapu 1 , Antonett Madriaga 1 , Aditya S Vaidya 1 , Irida Kastrati 2 , Bhargava Karumudi 1 , Yue-Ting Wang 1 , Taha Y Taha 1 , Gregory R J Thatcher 1 , Jonna Frasor 2 , Pavel A Petukhov 1
ChemMedChem ( IF 3.6 ) Pub Date : 2017-11-30 , DOI: 10.1002/cmdc.201700449 Hazem Abdelkarim 1 , Raghupathi Neelarapu 1 , Antonett Madriaga 1 , Aditya S Vaidya 1 , Irida Kastrati 2 , Bhargava Karumudi 1 , Yue-Ting Wang 1 , Taha Y Taha 1 , Gregory R J Thatcher 1 , Jonna Frasor 2 , Pavel A Petukhov 1
Affiliation
|
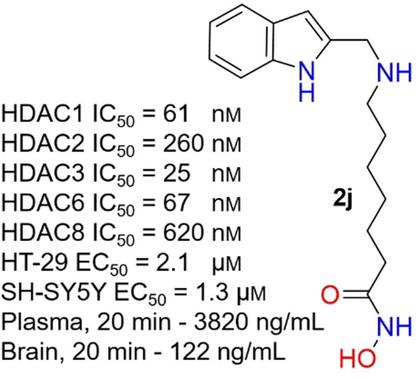
Histone deacetylases (HDACs) are promising drug targets for a variety of therapeutic applications. Herein we describe the design, synthesis, biological evaluation in cellular models of cancer, and preliminary drug metabolism and pharmacokinetic studies (DMPK) of a series of secondary and tertiary N‐substituted 7‐aminoheptanohydroxamic acid‐based HDAC inhibitors. Introduction of an amino group with one or two surface binding groups (SBGs) yielded a successful strategy to develop novel and potent HDAC inhibitors. The secondary amines were found to be generally more potent than the corresponding tertiary amines. Docking studies suggested that the SBGs of tertiary amines cannot be favorably accommodated at the gorge region of the binding site. The secondary amines with naphthalen‐2‐ylmethyl, 5‐phenylthiophen‐2‐ylmethyl, and 1H‐indol‐2‐ylmethyl (2 j) substituents exhibited the highest potency against class I HDACs: HDAC1 IC50 39–61 nm, HDAC2 IC50 260–690 nm, HDAC3 IC50 25–68 nm, and HDAC8 IC50 320–620 nm. The cytotoxicity of a representative set of secondary and tertiary N‐substituted 7‐aminoheptanoic acid hydroxyamide‐based inhibitors against HT‐29, SH‐SY5Y, and MCF‐7 cancer cells correlated with their inhibition of HDAC1, 2, and 3 and was found to be similar to or better than that of suberoylanilide hydroxamic acid (SAHA). Compounds in this series increased the acetylation of histones H3 and H4 in a time‐dependent manner. DMPK studies indicated that secondary amine 2 j is metabolically stable and has plasma and brain concentrations >23‐ and >1.6‐fold higher than the IC50 value for class I HDACs, respectively. Overall, the secondary and tertiary N‐substituted 7‐aminoheptanoic acid hydroxyamide‐based inhibitors exhibit excellent lead‐ and drug‐like properties and therapeutic capacity for cancer applications.
中文翻译:

新型胺基组蛋白脱乙酰酶抑制剂的设计、合成、分子建模和生物学评价
组蛋白脱乙酰酶 (HDAC) 是多种治疗应用的有前景的药物靶点。在此,我们描述了一系列二级和三级 N 取代 7-氨基庚异羟肟酸基 HDAC 抑制剂的设计、合成、癌症细胞模型中的生物学评价以及初步药物代谢和药代动力学研究 (DMPK)。引入具有一个或两个表面结合基团 (SBG) 的氨基为开发新型有效的 HDAC 抑制剂提供了成功的策略。发现仲胺通常比相应的叔胺更有效。对接研究表明,叔胺的SBG不能顺利地容纳在结合位点的峡谷区域。具有萘-2-基甲基、5-苯基噻吩-2-基甲基和 1 H-吲哚-2-基甲基 ( 2 j ) 取代基的仲胺对 I 类 HDAC 表现出最高的效力:HDAC1 IC 50 39-61 nm , HDAC2 IC 50 260–690 nm 、HDAC3 IC 50 25–68 nm和 HDAC8 IC 50 320–620 nm 。一组代表性的二级和三级 N 取代 7-氨基庚酸羟基酰胺抑制剂对 HT-29、SH-SY5Y 和 MCF-7 癌细胞的细胞毒性与其对 HDAC1、2 和 3 的抑制相关,并被发现类似于或优于辛二酰苯胺异羟肟酸(SAHA)。该系列化合物以时间依赖性方式增加组蛋白 H3 和 H4 的乙酰化。 DMPK 研究表明,仲胺2 j代谢稳定,血浆和脑浓度 >23- 和 >1。分别比 I 类 HDAC 的 IC 50值高 6 倍。总体而言,基于 N 取代的 7-氨基庚酸羟基酰胺的二级和三级抑制剂在癌症应用中表现出优异的先导和类药特性以及治疗能力。
更新日期:2017-11-30
中文翻译:
新型胺基组蛋白脱乙酰酶抑制剂的设计、合成、分子建模和生物学评价
组蛋白脱乙酰酶 (HDAC) 是多种治疗应用的有前景的药物靶点。在此,我们描述了一系列二级和三级 N 取代 7-氨基庚异羟肟酸基 HDAC 抑制剂的设计、合成、癌症细胞模型中的生物学评价以及初步药物代谢和药代动力学研究 (DMPK)。引入具有一个或两个表面结合基团 (SBG) 的氨基为开发新型有效的 HDAC 抑制剂提供了成功的策略。发现仲胺通常比相应的叔胺更有效。对接研究表明,叔胺的SBG不能顺利地容纳在结合位点的峡谷区域。具有萘-2-基甲基、5-苯基噻吩-2-基甲基和 1 H-吲哚-2-基甲基 ( 2 j ) 取代基的仲胺对 I 类 HDAC 表现出最高的效力:HDAC1 IC 50 39-61 nm , HDAC2 IC 50 260–690 nm 、HDAC3 IC 50 25–68 nm和 HDAC8 IC 50 320–620 nm 。一组代表性的二级和三级 N 取代 7-氨基庚酸羟基酰胺抑制剂对 HT-29、SH-SY5Y 和 MCF-7 癌细胞的细胞毒性与其对 HDAC1、2 和 3 的抑制相关,并被发现类似于或优于辛二酰苯胺异羟肟酸(SAHA)。该系列化合物以时间依赖性方式增加组蛋白 H3 和 H4 的乙酰化。 DMPK 研究表明,仲胺2 j代谢稳定,血浆和脑浓度 >23- 和 >1。分别比 I 类 HDAC 的 IC 50值高 6 倍。总体而言,基于 N 取代的 7-氨基庚酸羟基酰胺的二级和三级抑制剂在癌症应用中表现出优异的先导和类药特性以及治疗能力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号