Catalysis Today ( IF 5.2 ) Pub Date : 2017-10-26 , DOI: 10.1016/j.cattod.2017.10.040 Sijia Ding , Yasong Zhou , Qiang Wei , Shujiao Jiang , Wenwu Zhou
|
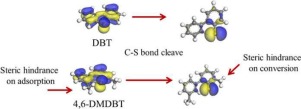
To study the substituent effects of 4,6-dimethyl-dibenzothiophene (4,6-DMDBT) on the direct hydrodesulfurization (DDS) routes catalyzed on a Ni-Mo-S nanocluster, a non-periodic computational Ni-Mo-S model is established, and density functional theory (DFT) is used to comparatively calculate the adsorption and the conversion of the dibenzothiophene (DBT) and 4,6-DMDBT on the Ni-S-edge, Ni-Mo-edge and corner sites of the Ni-Mo-S active nanocluster. The calculation results show that on the Ni-Mo-S active nanocluster, the Ni-S-edge could stably provide active hydrogen, whereas the CS bond cleavage on this active site required higher activation energy. The Ni-Mo-edge could not stably provide active hydrogen, and this active site has the highest activity of C
S bond cleavage. The corner site shows favorable ability of hydrogen activation and C
S bond cleavage. The methyl group of 4,6-DMDBT weakens the Ni
S bonds caused by the adsorption on the Ni-S-edge, where the decrease of the adsorption energy is compensated by an additional dispersion provided by two methyl groups. The two methyl groups also decrease the adsorption angle between the 4,6-DMDBT and the CUS sites on the Ni-Mo-edge and corner sites. The flat adsorption of 4,6-DMDBT will further hinder the hydrogen activation and transfer on the Ni-Mo-edge. Moreover, the methyl group of 4,6-DMDBT decreases the energy difference between the reactant and the intermediate of the DDS route, increasing the reaction energy of the C
S bond cleavage. The dominant DDS route of the DBT and 4,6-DMDBT involves the interaction between active hydrogen and the α-C followed by the movement of the newly formed aromatic ring away from the S atom of the sulfur compounds. During this process, the methyl groups of the 4,6-DMDBT will hinder the C
S bond cleavage on all CUS sites on the Ni-S-edge, Ni-Mo-edge and corner sites of the Ni-Mo-S nanocluster. Therefore, the activation energy of the C
S bond cleavage of 4,6-DMDBT is higher than that of DBT, and this is the main reason for the lower DDS rate of the former compared to the latter.
中文翻译:
4,6-DMDBT对Ni-Mo-S活性纳米团簇催化直接加氢脱硫路线的取代作用-理论研究
为了研究4,6-二甲基-二苯并噻吩(4,6-DMDBT)在Ni-Mo-S纳米团簇上催化的直接加氢脱硫(DDS)路线上的取代基效应,采用非周期性计算的Ni-Mo-S模型建立并使用密度泛函理论(DFT)来比较计算二苯并噻吩(DBT)和4,6-DMDBT在Ni-S边缘,Ni-Mo边缘和Ni拐角部位的吸附和转化-Mo-S活性纳米簇。计算结果表明,在Ni-Mo-S活性纳米簇上,Ni-S-edge可以稳定地提供活性氢,而在该活性位点上的C S键断裂需要更高的活化能。Ni-Mo-edge无法稳定地提供活性氢,并且该活性位点具有最高的C活性
S键裂解。拐角部位显示出有利的氢活化和
CS键裂解的能力。4,6-DMDBT的甲基弱化了
由于在Ni-S边缘上的吸附而引起的Ni S键,其中吸附能的下降被两个甲基提供的额外分散所补偿。两个甲基还降低了4,6-DMDBT与Ni-Mo边缘和拐角部位CUS部位之间的吸附角。4,6-DMDBT的平整吸附将进一步阻碍氢的活化和在Ni-Mo边缘的转移。此外,4,6-DMDBT的甲基减少了反应物与DDS路线中间体之间的能量差,从而增加了C的反应能。
S键裂解。DBT和4,6-DMDBT的主要DDS路线涉及活性氢与α-C之间的相互作用,然后是新形成的芳环从硫化合物的S原子移开。在此过程中,4,6-DMDBT的甲基将阻碍
在Ni-Mo-S纳米簇的Ni-S边缘,Ni-Mo边缘和角落位置的所有CUS位点上的C S键断裂。因此,
4,6-DMDBT的C S键断裂的活化能比DBT高,这是前者的DDS速率比后者低的主要原因。










































 京公网安备 11010802027423号
京公网安备 11010802027423号