当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Interaction entropy for computational alanine scanning in protein–protein binding
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2017-10-24 , DOI: 10.1002/wcms.1342 Linqiong Qiu 1 , Yuna Yan 1 , Zhaoxi Sun 1 , Jianing Song 1, 2 , John Z.H. Zhang 1, 2, 3, 4
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2017-10-24 , DOI: 10.1002/wcms.1342 Linqiong Qiu 1 , Yuna Yan 1 , Zhaoxi Sun 1 , Jianing Song 1, 2 , John Z.H. Zhang 1, 2, 3, 4
Affiliation
|
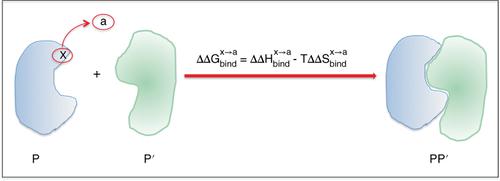
Protein–protein interactions (PPIs) are at the heart of signal transduction and are central to the function of protein machine in biology. The highly specific protein–protein binding is quantitatively characterized by the binding free energy whose accurate calculation from first principle is a grand challenge in computational biology. Accurate prediction of critical residues along with their specific and quantitative contributions to protein–protein binding free energy is extremely helpful to reveal binding mechanisms and identify drug‐like molecules that alter PPIs. In this overview, we describe an interaction entropy (IE) approach combined with the MM/GBSA method for solvation to compute residue‐specific protein–protein binding free energy. In this approach, the entropic contribution to binding free energy of individual residue is explicitly computed by using the IE method from a single MD trajectory. Studies for an extensive set of realistic protein–protein interaction systems demonstrated that by including the entropic contribution, the agreement between the computed residue‐specific binding free energies and the corresponding experimental data is systematically improved. We also show application of the current approach to the important major histocompatibility complex (MHC)‐antigen binding to provide important information on hot spots with potential application for use in cancer vaccine. WIREs Comput Mol Sci 2018, 8:e1342. doi: 10.1002/wcms.1342
中文翻译:

相互作用熵用于蛋白质-蛋白质结合中的计算丙氨酸扫描
蛋白质间相互作用(PPI)是信号转导的核心,对于生物学中蛋白质机器的功能至关重要。高度特异性的蛋白质-蛋白质结合的特征在于结合自由能,其从第一原理的精确计算是计算生物学中的巨大挑战。准确预测关键残基以及它们对蛋白质-蛋白质结合自由能的特定和定量贡献,对于揭示结合机制和识别可改变PPI的类药物分子非常有用。在本概述中,我们描述了一种相互作用熵(IE)方法与MM / GBSA方法相结合进行溶剂化,以计算残基特异性蛋白与蛋白结合的自由能。用这种方法 通过使用IE方法从单个MD轨迹显式计算熵对单个残基结合自由能的贡献。对大量现实的蛋白质-蛋白质相互作用系统的研究表明,通过包括熵的贡献,系统地改善了计算的残基特异性结合自由能与相应实验数据之间的一致性。我们还展示了当前方法在重要的主要组织相容性复合体(MHC)-抗原结合中的应用,从而为热点提供了重要信息,并有望用于癌症疫苗。系统地改善了计算的残基特异性结合自由能与相应实验数据之间的一致性。我们还展示了当前方法在重要的主要组织相容性复合体(MHC)-抗原结合中的应用,从而为热点提供了重要信息,并有望用于癌症疫苗。系统地改善了计算的残基特异性结合自由能与相应实验数据之间的一致性。我们还展示了当前方法在重要的主要组织相容性复合体(MHC)-抗原结合中的应用,从而为热点提供了重要信息,并有望用于癌症疫苗。电线计算科学2018,8:e1342。doi:10.1002 / wcms.1342
更新日期:2017-10-24
中文翻译:
相互作用熵用于蛋白质-蛋白质结合中的计算丙氨酸扫描
蛋白质间相互作用(PPI)是信号转导的核心,对于生物学中蛋白质机器的功能至关重要。高度特异性的蛋白质-蛋白质结合的特征在于结合自由能,其从第一原理的精确计算是计算生物学中的巨大挑战。准确预测关键残基以及它们对蛋白质-蛋白质结合自由能的特定和定量贡献,对于揭示结合机制和识别可改变PPI的类药物分子非常有用。在本概述中,我们描述了一种相互作用熵(IE)方法与MM / GBSA方法相结合进行溶剂化,以计算残基特异性蛋白与蛋白结合的自由能。用这种方法 通过使用IE方法从单个MD轨迹显式计算熵对单个残基结合自由能的贡献。对大量现实的蛋白质-蛋白质相互作用系统的研究表明,通过包括熵的贡献,系统地改善了计算的残基特异性结合自由能与相应实验数据之间的一致性。我们还展示了当前方法在重要的主要组织相容性复合体(MHC)-抗原结合中的应用,从而为热点提供了重要信息,并有望用于癌症疫苗。系统地改善了计算的残基特异性结合自由能与相应实验数据之间的一致性。我们还展示了当前方法在重要的主要组织相容性复合体(MHC)-抗原结合中的应用,从而为热点提供了重要信息,并有望用于癌症疫苗。系统地改善了计算的残基特异性结合自由能与相应实验数据之间的一致性。我们还展示了当前方法在重要的主要组织相容性复合体(MHC)-抗原结合中的应用,从而为热点提供了重要信息,并有望用于癌症疫苗。电线计算科学2018,8:e1342。doi:10.1002 / wcms.1342










































 京公网安备 11010802027423号
京公网安备 11010802027423号