PLoS Pathogens ( IF 5.5 ) Pub Date : 2017-10-19 , DOI: 10.1371/journal.ppat.1006644 Muhamuda Kader , Mounia Alaoui-EL-Azher , Jennie Vorhauer , Bhushan B Kode , Jakob Z. Wells , Donna Stolz , George Michalopoulos , Alan Wells , Melanie Scott , Nahed Ismail
|
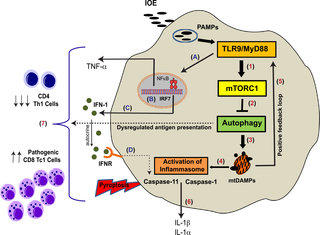
Severe hepatic inflammation is a common cause of acute liver injury following systemic infection with Ehrlichia, obligate Gram-negative intracellular bacteria that lack lipopolysaccharide (LPS). We have previously shown that type I IFN (IFN-I) and inflammasome activation are key host-pathogenic mediators that promote excessive inflammation and liver damage following fatal Ehrlichia infection. However, the underlying signals and mechanisms that regulate protective immunity and immunopathology during Ehrlichia infection are not well understood. To address this issue, we compared susceptibility to lethal Ixodes ovatus Ehrlichia (IOE) infection between wild type (WT) and MyD88-deficient (MyD88-/-) mice. We show here that MyD88-/- mice exhibited decreased inflammasome activation, attenuated liver injury, and were more resistant to lethal infection than WT mice, despite suppressed protective immunity and increased bacterial burden in the liver. MyD88-dependent inflammasome activation was also dependent on activation of the metabolic checkpoint kinase mammalian target of rapamycin complex 1 (mTORC1), inhibition of autophagic flux, and defective mitophagy in macrophages. Blocking mTORC1 signaling in infected WT mice and primary macrophages enhanced bacterial replication and attenuated inflammasome activation, suggesting autophagy promotes bacterial replication while inhibiting inflammasome activation. Finally, our data suggest TLR9 and IFN-I are upstream signaling mechanisms triggering MyD88-mediated mTORC1 and inflammasome activation in macrophages following Ehrlichia infection. This study reveals that Ehrlichia-induced liver injury and toxic shock are mediated by MyD88-dependent inflammasome activation and autophagy inhibition.
中文翻译:
依赖MyD88的炎症小体激活和自噬抑制作用导致衣原体诱导的肝损伤和中毒性休克
严重的肝炎是全身感染埃希氏菌(一种缺乏脂多糖(LPS)的革兰氏阴性细胞内细菌)引起的急性肝损伤的常见原因。先前我们已经表明,I型干扰素(IFN-I)和炎性体激活是关键的宿主致病性介质,可在致命的埃希氏菌感染后促进过度的炎症和肝脏损害。然而,目前尚不清楚在沙眼衣原体感染期间调节保护性免疫和免疫病理的潜在信号和机制。为了解决这个问题,我们比较易患致命的硬蜱卵形埃立克体(IOE)野生型之间感染(WT)和MyD88的缺陷(MyD88的- / -) 老鼠。我们在这里显示MyD88 -/-尽管保护性免疫力降低和肝脏细菌负荷增加,小鼠比WT小鼠表现出减少的炎性体活化,减轻的肝损伤和对致命感染的抵抗力。MyD88依赖性炎症小体激活还取决于雷帕霉素复合物1(mTORC1)的代谢检查点激酶哺乳动物靶标的激活,自噬通量的抑制和巨噬细胞中的吞噬缺陷。在受感染的野生型小鼠和原代巨噬细胞中阻断mTORC1信号传导可增强细菌复制并减弱炎症小体活化,表明自噬可促进细菌复制,同时抑制炎症小体活化。最后,我们的数据表明TLR9和IFN-I是触发MyD88介导的mTORC1和继发于巨噬细胞的炎性体激活的上游信号传导机制。埃希氏菌感染。这项研究表明,衣原体诱导的肝损伤和中毒性休克是由MyD88依赖性炎性体激活和自噬抑制介导的。










































 京公网安备 11010802027423号
京公网安备 11010802027423号