Journal of Catalysis ( IF 6.5 ) Pub Date : 2017-10-13 , DOI: 10.1016/j.jcat.2017.09.008 Genkuo Nie , Guozhu Li , Decheng Liang , Xiangwen Zhang
|
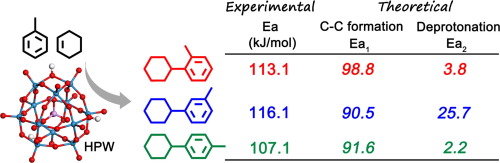
CC bond formation between an aromatic ring and cyclohexene is of great importance for the preparation of bicyclic hydrocarbons. In this work, alkylation of toluene with cyclohexene over phosphotungstic acid (HPW) has been investigated using both experimental reaction kinetics and ab initio density functional theory (DFT) methods. Kinetic models for the formations of o-cyclohexyltoluene, m-cyclohexyltoluene and p-cyclohexyltoluene have been established and used to fit the experimental kinetic data. The reaction rate constants and apparent activation energies for the formation of the three products have been calculated and compared. Based on DFT calculation, the energy profiles with relevant equilibrium and transition states have been determined for the adsorption of reactants, formation & deprotonation of the metastable states and desorption of the products. A metastable state of an adsorbed arenium ion on HPW has been demonstrated. The rate constants (k) for the major elementary steps at different temperatures have been calculated using DFT data. The apparent rate constants are further determined theoretically based on kinetic analysis. Results show that the theoretical calculation fits well with experimental data, both of which have similar tendency changing with temperature. Combined experimental kinetic results and DFT calculations suggest that both the formation and deprotonation of the metastable state play key roles in affecting the product selectivity.
中文翻译:
磷钨酸上甲苯与环己烯的烷基化:联合实验和计算研究
在芳环和环己烯之间形成C C键对于制备双环烃非常重要。在这项工作中,已使用实验反应动力学和从头算密度泛函理论(DFT)方法研究了磷化钨酸(HPW)上甲苯与环己烯的烷基化反应。动力学模型的地层Ó -cyclohexyltoluene,米-cyclohexyltoluene和p已经建立了β-环己基甲苯并将其用于拟合实验动力学数据。计算并比较了形成这三种产物的反应速率常数和表观活化能。基于DFT计算,已确定了具有相关平衡态和过渡态的能量分布图,用于反应物的吸附,亚稳态的形成和去质子化以及产物的解吸。已经证明了在HPW上吸附的芳烃离子处于亚稳态。速率常数(k),使用DFT数据计算出了不同温度下的主要基本步骤。在理论上基于动力学分析进一步确定表观速率常数。结果表明,理论计算与实验数据吻合良好,二者随温度的变化趋势相似。结合实验动力学结果和DFT计算表明,亚稳态的形成和去质子化在影响产物选择性方面起着关键作用。










































 京公网安备 11010802027423号
京公网安备 11010802027423号