当前位置:
X-MOL 学术
›
Chem. Soc. Rev.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theories and simulations of roaming
Chemical Society Reviews ( IF 40.4 ) Pub Date : 2017-10-05 00:00:00 , DOI: 10.1039/c7cs00578d Joel M. Bowman 1, 2, 3, 4 , Paul L. Houston 4, 5, 6, 7, 8
Chemical Society Reviews ( IF 40.4 ) Pub Date : 2017-10-05 00:00:00 , DOI: 10.1039/c7cs00578d Joel M. Bowman 1, 2, 3, 4 , Paul L. Houston 4, 5, 6, 7, 8
Affiliation
|
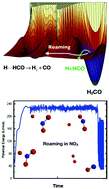
The phenomenon of roaming in chemical reactions has now become both commonly observed in experiment and extensively supported by theory and simulations. Roaming occurs in highly-excited molecules when the trajectories of atomic motion often bypass the minimum energy pathway and produce reaction in unexpected ways from unlikely geometries. The prototypical example is the unimolecular dissociation of formaldehyde (H2CO), in which the “normal” reaction proceeds through a tight transition state to yield H2 + CO but for which a high fraction of dissociations take place via a “roaming” mechanism in which one H atom moves far from the HCO, almost to dissociation, and then returns to abstract the second H atom. We review below the theories and simulations that have recently been developed to address and understand this new reaction phenomenon.
中文翻译:

漫游理论与模拟
现在,化学反应中的漫游现象已成为实验中普遍观察到的现象,并已得到理论和模拟的广泛支持。当原子运动的轨迹经常绕过最小能量路径并以不太可能的几何形状以意想不到的方式产生反应时,漫游会发生在高度激发的分子中。典型的例子是甲醛(H 2 CO)的单分子解离,其中“正常”反应通过紧密的过渡态进行反应,生成H 2 + CO,但其中高比例的解离通过一种“漫游”机制,其中一个H原子远离HCO,几乎解离,然后返回以抽象出第二个H原子。我们在下面回顾了最近为解决和理解这种新的反应现象而开发的理论和模拟。
更新日期:2017-10-05
中文翻译:
漫游理论与模拟
现在,化学反应中的漫游现象已成为实验中普遍观察到的现象,并已得到理论和模拟的广泛支持。当原子运动的轨迹经常绕过最小能量路径并以不太可能的几何形状以意想不到的方式产生反应时,漫游会发生在高度激发的分子中。典型的例子是甲醛(H 2 CO)的单分子解离,其中“正常”反应通过紧密的过渡态进行反应,生成H 2 + CO,但其中高比例的解离通过一种“漫游”机制,其中一个H原子远离HCO,几乎解离,然后返回以抽象出第二个H原子。我们在下面回顾了最近为解决和理解这种新的反应现象而开发的理论和模拟。










































 京公网安备 11010802027423号
京公网安备 11010802027423号