当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theory and applications of surface micro‐kinetics in the rational design of catalysts using density functional theory calculations
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2017-06-05 , DOI: 10.1002/wcms.1321 Yu Mao 1, 2 , Hai‐Feng Wang 1 , P. Hu 1, 2
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2017-06-05 , DOI: 10.1002/wcms.1321 Yu Mao 1, 2 , Hai‐Feng Wang 1 , P. Hu 1, 2
Affiliation
|
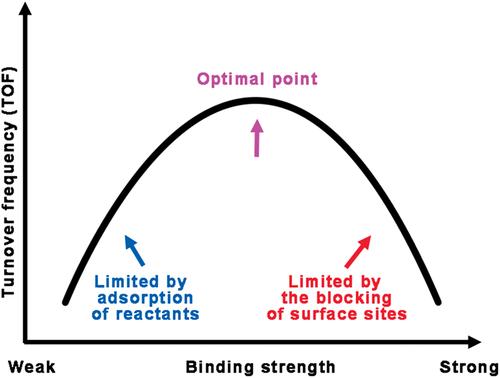
Rational design of catalysts has long been an important and challenging goal in heterogeneous catalysis. To achieve this target, density functional theory (DFT) calculations and micro‐kinetics are two of the cornerstones. The DFT calculations make it possible to obtain microscopic properties of catalytic systems by computational simulations, and the micro‐kinetic modeling of surface reactions provides a tool to link quantum‐chemical data with macroscopic behaviors of the systems. In this review, we focus on the basic concepts and latest theoretical progresses of strategies for the catalysts design, including Brønsted−Evans−Polanyi relation, the volcano curve, and the activity window. Among the progresses, the theory of chemical potential kinetics in heterogeneous catalysis and its implications on catalysts design, which was developed by our group, are described in detail with extensive derivations. Furthermore, the applications of this method on screening low‐cost counter electrodes for dye‐sensitized solar cells are presented with experimental evidences. WIREs Comput Mol Sci 2017, 7:e1321. doi: 10.1002/wcms.1321
中文翻译:

使用密度泛函理论计算的表面微动力学理论在催化剂合理设计中的应用
长期以来,合理设计催化剂一直是非均相催化中一个重要且具有挑战性的目标。为了实现这一目标,密度泛函理论(DFT)计算和微动力学是两个基石。DFT计算使通过计算模拟获得催化系统的微观特性成为可能,表面反应的微观动力学模型提供了将量子化学数据与系统宏观行为联系起来的工具。在这篇综述中,我们关注于催化剂设计策略的基本概念和最新理论进展,包括布朗斯台德-埃文斯-波兰尼关系,火山曲线和活动窗口。在研究进展中,我们小组开发了非均相催化中的化学势动力学理论及其对催化剂设计的影响,详细描述了广泛的派生。此外,该方法在筛选染料敏化太阳能电池的低成本对电极中的应用具有实验证据。电线Comput Mol Sci 2017,7:e1321。doi:10.1002 / wcms.1321
更新日期:2017-06-05
中文翻译:
使用密度泛函理论计算的表面微动力学理论在催化剂合理设计中的应用
长期以来,合理设计催化剂一直是非均相催化中一个重要且具有挑战性的目标。为了实现这一目标,密度泛函理论(DFT)计算和微动力学是两个基石。DFT计算使通过计算模拟获得催化系统的微观特性成为可能,表面反应的微观动力学模型提供了将量子化学数据与系统宏观行为联系起来的工具。在这篇综述中,我们关注于催化剂设计策略的基本概念和最新理论进展,包括布朗斯台德-埃文斯-波兰尼关系,火山曲线和活动窗口。在研究进展中,我们小组开发了非均相催化中的化学势动力学理论及其对催化剂设计的影响,详细描述了广泛的派生。此外,该方法在筛选染料敏化太阳能电池的低成本对电极中的应用具有实验证据。电线Comput Mol Sci 2017,7:e1321。doi:10.1002 / wcms.1321










































 京公网安备 11010802027423号
京公网安备 11010802027423号