当前位置:
X-MOL 学术
›
Faraday Discuss.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Design principles from multiscale simulations to predict nanostructure in self-assembling ionic liquids
Faraday Discussions ( IF 3.4 ) Pub Date : 2017-07-18 00:00:00 , DOI: 10.1039/c7fd00154a Benjamin T. Nebgen 1, 2, 3, 4, 5 , Harsha D. Magurudeniya 1, 2, 3, 4, 5 , Kevin W. C. Kwock 1, 2, 3, 4, 5 , Bryan S. Ringstrand 1, 2, 3, 4, 5 , Towfiq Ahmed 2, 3, 4, 5, 6 , Sönke Seifert 7, 8, 9, 10 , Jian-Xin Zhu 1, 2, 3, 4, 5 , Sergei Tretiak 1, 2, 3, 4, 5 , Millicent A. Firestone 1, 2, 3, 4, 5
Faraday Discussions ( IF 3.4 ) Pub Date : 2017-07-18 00:00:00 , DOI: 10.1039/c7fd00154a Benjamin T. Nebgen 1, 2, 3, 4, 5 , Harsha D. Magurudeniya 1, 2, 3, 4, 5 , Kevin W. C. Kwock 1, 2, 3, 4, 5 , Bryan S. Ringstrand 1, 2, 3, 4, 5 , Towfiq Ahmed 2, 3, 4, 5, 6 , Sönke Seifert 7, 8, 9, 10 , Jian-Xin Zhu 1, 2, 3, 4, 5 , Sergei Tretiak 1, 2, 3, 4, 5 , Millicent A. Firestone 1, 2, 3, 4, 5
Affiliation
|
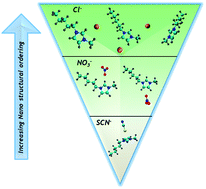
Molecular dynamics simulations (up to the nanoscale) were performed on the 3-methyl-1-pentylimidazolium ionic liquid cation paired with three anions; chloride, nitrate, and thiocyanate as aqueous mixtures, using the effective fragment potential (EFP) method, a computationally inexpensive way of modeling intermolecular interactions. The simulations provided insight (preferred geometries, radial distribution functions and theoretical proton NMR resonances) into the interactions within the ionic domain and are validated against 1H NMR spectroscopy and small- and wide-angle X-ray scattering experiments on 1-decyl-3-methylimidazolium. Ionic liquids containing thiocyanate typically resist gelation and form poorly ordered lamellar structures upon mixing with water. Conversely, chloride, a strongly coordinating anion, normally forms strong physical gels and produces well-ordered nanostructures adopting a variety of structural motifs over a very wide range of water compositions. Nitrate is intermediate in character, whereby upon dispersal in water it displays a range of viscosities and self-assembles into nanostructures with considerable variability in the fidelity of ordering and symmetry, as a function of water content in the binary mixtures. The observed changes in the macro and nanoscale characteristics were directly correlated to ionic domain structures and intermolecular interactions as theoretically predicted by the analysis of MD trajectories and calculated RDFs. Specifically, both chloride and nitrate are positioned in the plane of the cation. Anion to cation proximity is dependent on water content. Thiocyanate is more susceptible to water insertion into the second solvent shell. Experimental 1H NMR chemical shifts monitor the site-specific competition dependence with water content in the binary mixtures. Thiocyanate preferentially sits above and below the aromatic ring plane, a state disallowing interaction with the protons on the imidazolium ring.
中文翻译:

从多尺度模拟中预测自组装离子液体中纳米结构的设计原理
对与三个阴离子配对的3-甲基-1-戊咪唑鎓离子液体阳离子进行了分子动力学模拟(达到纳米级)。氯化物,硝酸盐和硫氰酸盐作为水性混合物,使用有效片段电位(EFP)方法进行计算,该方法是计算分子间相互作用的廉价计算方法。模拟为离子域内的相互作用提供了洞察力(首选的几何形状,径向分布函数和理论质子NMR共振),并针对1进行了验证。1癸基-3-甲基咪唑鎓的1 H NMR光谱学和小角度和广角X射线散射实验。含有硫氰酸盐的离子液体通常会抵抗胶凝作用,并在与水混合后形成排列不规则的层状结构。相反,氯离子是一种强配位阴离子,通常会形成强力的物理凝胶,并产生排列整齐的纳米结构,这些结构在很宽的水组成范围内采用了多种结构图案。硝酸盐的性质是中等的,因此分散在水中后,它显示出一定范围的粘度,并根据二元混合物中水含量的变化,自组装成纳米结构,其保真度和对称性变化很大。所观察到的宏观和纳米尺度特征的变化与离子域结构和分子间相互作用直接相关,正如通过MD轨迹分析和计算得出的RDF所理论预测的那样。具体地说,氯化物和硝酸盐都位于阳离子平面内。阴离子与阳离子的接近度取决于水含量。硫氰酸盐更容易将水插入第二溶剂壳中。实验性1 H NMR化学位移监测二元混合物中水的含量与位点竞争的依赖性。硫氰酸盐优先位于芳香环平面的上方和下方,这种状态不允许与咪唑环上的质子发生相互作用。
更新日期:2017-12-15
中文翻译:
从多尺度模拟中预测自组装离子液体中纳米结构的设计原理
对与三个阴离子配对的3-甲基-1-戊咪唑鎓离子液体阳离子进行了分子动力学模拟(达到纳米级)。氯化物,硝酸盐和硫氰酸盐作为水性混合物,使用有效片段电位(EFP)方法进行计算,该方法是计算分子间相互作用的廉价计算方法。模拟为离子域内的相互作用提供了洞察力(首选的几何形状,径向分布函数和理论质子NMR共振),并针对1进行了验证。1癸基-3-甲基咪唑鎓的1 H NMR光谱学和小角度和广角X射线散射实验。含有硫氰酸盐的离子液体通常会抵抗胶凝作用,并在与水混合后形成排列不规则的层状结构。相反,氯离子是一种强配位阴离子,通常会形成强力的物理凝胶,并产生排列整齐的纳米结构,这些结构在很宽的水组成范围内采用了多种结构图案。硝酸盐的性质是中等的,因此分散在水中后,它显示出一定范围的粘度,并根据二元混合物中水含量的变化,自组装成纳米结构,其保真度和对称性变化很大。所观察到的宏观和纳米尺度特征的变化与离子域结构和分子间相互作用直接相关,正如通过MD轨迹分析和计算得出的RDF所理论预测的那样。具体地说,氯化物和硝酸盐都位于阳离子平面内。阴离子与阳离子的接近度取决于水含量。硫氰酸盐更容易将水插入第二溶剂壳中。实验性1 H NMR化学位移监测二元混合物中水的含量与位点竞争的依赖性。硫氰酸盐优先位于芳香环平面的上方和下方,这种状态不允许与咪唑环上的质子发生相互作用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号