当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Multiconfigurational actinide nitrides assisted by double Möbius aromaticity
Chemical Science ( IF 8.4 ) Pub Date : 2024-04-26 , DOI: 10.1039/d4sc01549e Xuhui Lin 1 , Xiaoli Lu 2 , Shenghui Tang 2 , Wei Wu 3 , Yirong Mo 4
Chemical Science ( IF 8.4 ) Pub Date : 2024-04-26 , DOI: 10.1039/d4sc01549e Xuhui Lin 1 , Xiaoli Lu 2 , Shenghui Tang 2 , Wei Wu 3 , Yirong Mo 4
Affiliation
|
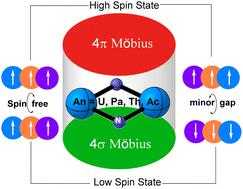
Understanding the bonding nature between actinides and main-group elements remains a key challenge in actinide chemistry due to the involvement of f orbitals. Herein, we propose a unique “aromaticity-assisted multiconfiguration” (AAM) model to elucidate the bonding nature in actinide nitrides (An2N2, An = Ac, Th, Pa, U). Each planar four-membered An2N2 with equivalent An–N bonds possesses four delocalized π electrons and four delocalized σ electrons, forming a new family of double Möbius aromaticity that contributes to the molecular stability. The unprecedented aromaticity further supports actinide nitrides to exhibit multiconfigurational characters, where the unpaired electrons (2, 4 or 6 in naked Th2N2, Pa2N2 or U2N2, respectively) either are spin-free and localized on metal centres or form metal–ligand bonds. High-level multiconfigurational computations confirm an open-shell singlet ground state for actinide nitrides, with small energy gaps to high spin states. This is consistent with the antiferromagnetic nature observed experimentally in uranium nitrides. The novel AAM bonding model can be authenticated in both experimentally identified compounds containing a U2N2 motif and other theoretically modelled An2N2 clusters and is thus expected to be a general chemical bonding pattern between actinides and main-group elements.
中文翻译:

双莫比乌斯芳香辅助的多构型锕系氮化物
由于 f 轨道的参与,了解锕系元素和主族元素之间的键合性质仍然是锕系化学中的一个关键挑战。在此,我们提出了一种独特的“芳香辅助多构型”(AAM)模型来阐明锕系氮化物中的键合性质(An 2 N 2,An = Ac,Th,Pa,U)。每个具有等效 An-N 键的平面四元 An 2 N 2拥有四个离域 π 电子和四个离域 σ 电子,形成一个新的双莫比乌斯芳香族,有助于分子稳定性。前所未有的芳香性进一步支持锕系氮化物表现出多构型特征,其中不成对电子(裸Th 2 N 2、Pa 2 N 2或U 2 N 2中分别有2、4或6个)要么是无自旋的,要么是局域在金属上中心或形成金属-配体键。高级多构型计算证实了锕系氮化物的开壳单线态基态,与高自旋态的能隙较小。这与在氮化铀中实验观察到的反铁磁性质一致。新的AAM键合模型可以在实验鉴定的含有U 2 N 2基序的化合物和其他理论模型的An 2 N 2簇中得到验证,因此有望成为锕系元素和主族元素之间的通用化学键合模式。
更新日期:2024-04-26
中文翻译:
双莫比乌斯芳香辅助的多构型锕系氮化物
由于 f 轨道的参与,了解锕系元素和主族元素之间的键合性质仍然是锕系化学中的一个关键挑战。在此,我们提出了一种独特的“芳香辅助多构型”(AAM)模型来阐明锕系氮化物中的键合性质(An 2 N 2,An = Ac,Th,Pa,U)。每个具有等效 An-N 键的平面四元 An 2 N 2拥有四个离域 π 电子和四个离域 σ 电子,形成一个新的双莫比乌斯芳香族,有助于分子稳定性。前所未有的芳香性进一步支持锕系氮化物表现出多构型特征,其中不成对电子(裸Th 2 N 2、Pa 2 N 2或U 2 N 2中分别有2、4或6个)要么是无自旋的,要么是局域在金属上中心或形成金属-配体键。高级多构型计算证实了锕系氮化物的开壳单线态基态,与高自旋态的能隙较小。这与在氮化铀中实验观察到的反铁磁性质一致。新的AAM键合模型可以在实验鉴定的含有U 2 N 2基序的化合物和其他理论模型的An 2 N 2簇中得到验证,因此有望成为锕系元素和主族元素之间的通用化学键合模式。


























 京公网安备 11010802027423号
京公网安备 11010802027423号