当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Efficient Exploitation of Numerical Quadrature with Distance-Dependent Integral Screening in Explicitly Correlated F12 Theory: Linear Scaling Evaluation of the Most Expensive RI-MP2-F12 Term
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2024-04-16 , DOI: 10.1021/acs.jctc.4c00193 Lars Urban 1, 2 , Henryk Laqua 1 , Travis H. Thompson 1 , Christian Ochsenfeld 1, 2
Journal of Chemical Theory and Computation ( IF 5.5 ) Pub Date : 2024-04-16 , DOI: 10.1021/acs.jctc.4c00193 Lars Urban 1, 2 , Henryk Laqua 1 , Travis H. Thompson 1 , Christian Ochsenfeld 1, 2
Affiliation
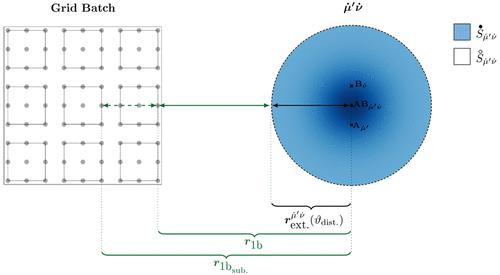
|
We present a linear scaling atomic orbital based algorithm for the computation of the most expensive exchange-type RI-MP2-F12 term by employing numerical quadrature in combination with CABS-RI to avoid six-center-three-electron integrals. Furthermore, a robust distance-dependent integral screening scheme, based on integral partition bounds [Thompson, T. H.; Ochsenfeld, C. J. Chem. Phys. 2019, 150, 044101], is used to drastically reduce the number of the required three-center-one-electron integrals substantially. The accuracy of our numerical quadrature/CABS-RI approach and the corresponding integral screening is thoroughly assessed for interaction and isomerization energies across a variety of numerical integration grids. Our method outperforms the standard density fitting/CABS-RI approach with errors below 1 μEh even for small grid sizes and moderate screening thresholds. The choice of the grid size and screening threshold allows us to tailor our ansatz to a desired accuracy and computational efficiency. We showcase the approach’s effectiveness for the chemically relevant system valinomycin, employing a triple-ζ F12 basis set combination (C54H90N6O18, 5757 AO basis functions, 10,266 CABS basis functions, 735,783 grid points). In this context, our ansatz achieves higher accuracy combined with a 135× speedup compared to the classical density fitting based variant, requiring notably less computation time than the corresponding RI-MP2 calculation. Additionally, we demonstrate near-linear scaling through calculations on linear alkanes. We achieved an 817-fold acceleration for C80H162 and an extrapolated 28,765-fold acceleration for C200H402, resulting in a substantially reduced computational time for the latter─from 229 days to just 11.5 min. Our ansatz may also be adapted to the remaining MP2-F12 terms, which will be the subject of future work.
中文翻译:

显式相关 F12 理论中距离相关积分筛选数值求积的有效利用:最昂贵 RI-MP2-F12 项的线性标度评估
我们提出了一种基于线性缩放原子轨道的算法,通过采用数值求积与 CABS-RI 相结合来计算最昂贵的交换型 RI-MP2-F12 项,以避免六中心三电子积分。此外,基于积分划分界限的稳健的距离相关积分筛选方案[Thompson,TH;奥克森菲尔德 (C. J. Chem)。物理。 2019, 150, 044101],用于大幅减少所需的三中心一电子积分的数量。我们的数值求积/CABS-RI 方法和相应的积分筛选的准确性经过彻底评估,以了解各种数值积分网格中的相互作用和异构化能。我们的方法优于标准密度拟合/CABS-RI 方法,即使对于小网格尺寸和中等筛选阈值,误差也低于 1 μE h 。网格大小和筛选阈值的选择使我们能够根据所需的精度和计算效率定制我们的 ansatz。我们展示了该方法对化学相关系统缬氨霉素的有效性,采用三重 z F12 基组组合(C 54 H 90 N 6 O 18、5757 AO 基函数、10,266 CABS 基函数、735,783 个网格点)。在这种情况下,与基于经典密度拟合的变体相比,我们的 ansatz 实现了更高的精度和 135 倍的加速,所需的计算时间明显少于相应的 RI-MP2 计算。此外,我们通过线性烷烃的计算证明了近线性缩放。我们对 C 80 H 162实现了 817 倍的加速,对 C 200 H 402实现了 28,765 倍的外推加速,从而大幅缩短了后者的计算时间 — 从 229 天缩短到仅 11.5 分钟。我们的 ansatz 也可能会适应剩余的 MP2-F12 条款,这将是未来工作的主题。
更新日期:2024-04-17
中文翻译:
显式相关 F12 理论中距离相关积分筛选数值求积的有效利用:最昂贵 RI-MP2-F12 项的线性标度评估
我们提出了一种基于线性缩放原子轨道的算法,通过采用数值求积与 CABS-RI 相结合来计算最昂贵的交换型 RI-MP2-F12 项,以避免六中心三电子积分。此外,基于积分划分界限的稳健的距离相关积分筛选方案[Thompson,TH;奥克森菲尔德 (C. J. Chem)。物理。 2019, 150, 044101],用于大幅减少所需的三中心一电子积分的数量。我们的数值求积/CABS-RI 方法和相应的积分筛选的准确性经过彻底评估,以了解各种数值积分网格中的相互作用和异构化能。我们的方法优于标准密度拟合/CABS-RI 方法,即使对于小网格尺寸和中等筛选阈值,误差也低于 1 μE h 。网格大小和筛选阈值的选择使我们能够根据所需的精度和计算效率定制我们的 ansatz。我们展示了该方法对化学相关系统缬氨霉素的有效性,采用三重 z F12 基组组合(C 54 H 90 N 6 O 18、5757 AO 基函数、10,266 CABS 基函数、735,783 个网格点)。在这种情况下,与基于经典密度拟合的变体相比,我们的 ansatz 实现了更高的精度和 135 倍的加速,所需的计算时间明显少于相应的 RI-MP2 计算。此外,我们通过线性烷烃的计算证明了近线性缩放。我们对 C 80 H 162实现了 817 倍的加速,对 C 200 H 402实现了 28,765 倍的外推加速,从而大幅缩短了后者的计算时间 — 从 229 天缩短到仅 11.5 分钟。我们的 ansatz 也可能会适应剩余的 MP2-F12 条款,这将是未来工作的主题。


























 京公网安备 11010802027423号
京公网安备 11010802027423号