Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Molecular Mechanisms of Sorbed Ion Effects during Boehmite Particle Aggregation
Langmuir ( IF 3.9 ) Pub Date : 2024-04-10 , DOI: 10.1021/acs.langmuir.3c03532 Tingting Liu 1 , Nikhil Rampal 1 , Elias Nakouzi 2 , Benjamin A. Legg 2 , Jaehun Chun 2 , Lili Liu 2 , Gregory K. Schenter 2 , James J. De Yoreo 2 , Lawrence M. Anovitz 1 , Andrew G. Stack 1
Langmuir ( IF 3.9 ) Pub Date : 2024-04-10 , DOI: 10.1021/acs.langmuir.3c03532 Tingting Liu 1 , Nikhil Rampal 1 , Elias Nakouzi 2 , Benjamin A. Legg 2 , Jaehun Chun 2 , Lili Liu 2 , Gregory K. Schenter 2 , James J. De Yoreo 2 , Lawrence M. Anovitz 1 , Andrew G. Stack 1
Affiliation
|
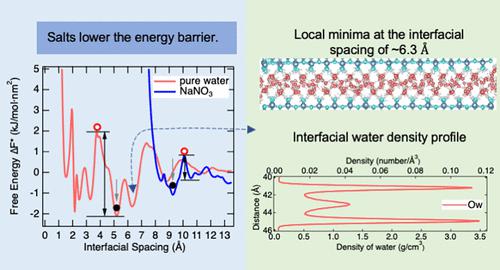
Classical theories of particle aggregation, such as Derjaguin–Landau–Verwey–Overbeek (DLVO), do not explain recent observations of ion-specific effects or the complex concentration dependence for aggregation. Thus, here, we probe the molecular mechanisms by which selected alkali nitrate ions (Na+, K+, and NO3–) influence aggregation of the mineral boehmite (γ-AlOOH) nanoparticles. Nanoparticle aggregation was analyzed using classical molecular dynamics (CMD) simulations coupled with the metadynamics rare event approach for stoichiometric surface terminations of two boehmite crystal faces. Calculated free energy landscapes reveal how electrolyte ions alter aggregation on different crystal faces relative to pure water. Consistent with experimental observations, we find that adding an electrolyte significantly reduces the energy barrier for particle aggregation (∼3–4×). However, in this work, we show this is due to the ions disrupting interstitial water networks, and that aggregation between stoichiometric (010) basal–basal surfaces is more favorable than between (001) edge–edge surfaces (∼5–6×) due to the higher interfacial water densities on edge surfaces. The interfacial distances in the interlayer between aggregated particles with electrolytes (∼5–10 Å) are larger than those in pure water (a few Ångströms). Together, aggregation/disaggregation in salt solutions is predicted to be more reversible due to these lower energy barriers, but there is uncertainty on the magnitudes of the energies that lead to aggregation at the molecular scale. By analyzing the peak water densities of the first monolayer of interstitial water as a proxy for solvent ordering, we find that the extent of solvent ordering likely determines the structures of aggregated states as well as the energy barriers to move between them. The results suggest a path for developing a molecular-level basis to predict the synergies between ions and crystal faces that facilitate aggregation under given solution conditions. Such fundamental understanding could be applied extensively to the aggregation and precipitation utilization in the biological, pharmaceutical, materials design, environmental remediation, and geological regimes.
中文翻译:

勃姆石颗粒聚集过程中吸附离子效应的分子机制
粒子聚集的经典理论,例如 Derjaguin–Landau–Verwey–Overbeek (DLVO),并不能解释最近观察到的离子特异性效应或聚集的复杂浓度依赖性。因此,在这里,我们探讨了选定的碱金属硝酸根离子(Na +、K +和 NO 3 -)影响矿物勃姆石(γ-AlOOH)纳米颗粒聚集的分子机制。使用经典分子动力学(CMD)模拟结合元动力学稀有事件方法对两个勃姆石晶面的化学计量表面终止分析纳米粒子聚集。计算的自由能景观揭示了电解质离子如何改变相对于纯水在不同晶面上的聚集。与实验观察一致,我们发现添加电解质显着降低了颗粒聚集的能垒(∼3-4×)。然而,在这项工作中,我们表明这是由于离子破坏了间隙水网络,并且化学计量的(010)基底-基底表面之间的聚集比(001)边缘-边缘表面之间的聚集更有利(∼5-6×)由于边缘表面的界面水密度较高。具有电解质的聚集颗粒之间的层间界面距离(〜5-10埃)比纯水中的界面距离(几埃)大。总之,由于这些较低的能垒,盐溶液中的聚集/解聚预计将更加可逆,但导致分子尺度聚集的能量大小存在不确定性。通过分析作为溶剂有序代理的第一单层间隙水的峰值水密度,我们发现溶剂有序的程度可能决定聚集态的结构以及它们之间移动的能量势垒。结果提出了一条开发分子水平基础的途径,以预测离子和晶面之间的协同作用,从而在给定的溶液条件下促进聚集。这种基本的理解可以广泛应用于生物、制药、材料设计、环境修复和地质状况中的聚集和沉淀利用。
更新日期:2024-04-10
中文翻译:
勃姆石颗粒聚集过程中吸附离子效应的分子机制
粒子聚集的经典理论,例如 Derjaguin–Landau–Verwey–Overbeek (DLVO),并不能解释最近观察到的离子特异性效应或聚集的复杂浓度依赖性。因此,在这里,我们探讨了选定的碱金属硝酸根离子(Na +、K +和 NO 3 -)影响矿物勃姆石(γ-AlOOH)纳米颗粒聚集的分子机制。使用经典分子动力学(CMD)模拟结合元动力学稀有事件方法对两个勃姆石晶面的化学计量表面终止分析纳米粒子聚集。计算的自由能景观揭示了电解质离子如何改变相对于纯水在不同晶面上的聚集。与实验观察一致,我们发现添加电解质显着降低了颗粒聚集的能垒(∼3-4×)。然而,在这项工作中,我们表明这是由于离子破坏了间隙水网络,并且化学计量的(010)基底-基底表面之间的聚集比(001)边缘-边缘表面之间的聚集更有利(∼5-6×)由于边缘表面的界面水密度较高。具有电解质的聚集颗粒之间的层间界面距离(〜5-10埃)比纯水中的界面距离(几埃)大。总之,由于这些较低的能垒,盐溶液中的聚集/解聚预计将更加可逆,但导致分子尺度聚集的能量大小存在不确定性。通过分析作为溶剂有序代理的第一单层间隙水的峰值水密度,我们发现溶剂有序的程度可能决定聚集态的结构以及它们之间移动的能量势垒。结果提出了一条开发分子水平基础的途径,以预测离子和晶面之间的协同作用,从而在给定的溶液条件下促进聚集。这种基本的理解可以广泛应用于生物、制药、材料设计、环境修复和地质状况中的聚集和沉淀利用。


























 京公网安备 11010802027423号
京公网安备 11010802027423号