Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Photocatalytic Efficiency for CO2 Reduction of Co and Cluster Co2O2 Supported on g-C3N4: A Density Functional Theory and Machine Learning Study
Langmuir ( IF 3.9 ) Pub Date : 2024-04-05 , DOI: 10.1021/acs.langmuir.3c03550 Elham Moharramzadeh Goliaei 1, 2, 3
Langmuir ( IF 3.9 ) Pub Date : 2024-04-05 , DOI: 10.1021/acs.langmuir.3c03550 Elham Moharramzadeh Goliaei 1, 2, 3
Affiliation
|
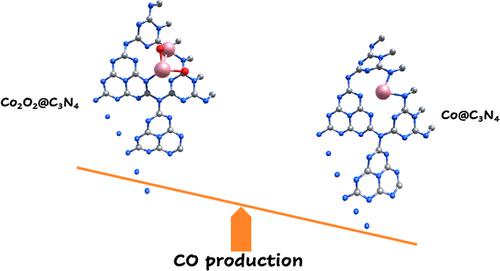
It is well known from experimental results that a single atom of cobalt supported on g-C3N4 is an efficient photocatalyst for the reduction of CO2 to CO, with a better photocatalytic activity than g-C3N4. In this work, we investigate the performance as catalysts for the CO2 reduction of single atoms of cobalt and Co2O2 clusters supported on graphitic carbon nitride (g-C3N4). Employing density functional theory plus Hubbard (DFT + U) calculations, we investigate in detail the reduction mechanisms to CO and HCOOH for the first time. We find that deposition of cobalt on g-C3N4 decreases the work function of g-C3N4 to 6.6 eV and provides a better candidate for the reduction reaction. In addition, we find that the preferred product of CO2 reduction on Co@g-C3N4 is CO, with a rate-determining barrier of 0.97 eV, while on Co2O2@g-C3N4, CO2 reduces to formate with a rate-determining barrier of 0.44 eV. We determine the creation of CO2 from COOH to only take place on Co2O2@g-C3N4, with a (relatively high) barrier of 2.27 eV. In order to obtain more easily the transition state energies of the reactions mentioned above, we applied machine learning methods to search for high-importance descriptors for these quantities, in the case of single transition metal atoms supported on C3N4. Interestingly, our results show that our quantities of interest are closely related to the adsorption energies of products and normalized valence electrons of the products of the elementary reactions as well as those of the metal atoms. The former of these two sets of features can be straightforwardly obtained via DFT, while the latter energies are extensively tabulated. Our results offer guidance for the design of catalysts and photocatalysts for CO2 reduction on single-metal atoms supported on C3N4.
中文翻译:

g-C3N4 负载的 Co 和团簇 Co2O2 光催化还原 CO2 效率:密度泛函理论和机器学习研究
从实验结果可知,gC 3 N 4负载的单原子钴是一种有效的将CO 2还原为CO的光催化剂,其光催化活性比gC 3 N 4更好。在这项工作中,我们研究了负载在石墨氮化碳(gC 3 N 4)上的钴和Co 2 O 2簇单原子的CO 2还原催化剂的性能。采用密度泛函理论加上 Hubbard (DFT + U ) 计算,我们首次详细研究了 CO 和 HCOOH 的还原机制。我们发现钴在gC 3 N 4上的沉积将gC 3 N 4的功函数降低至6.6 eV,并为还原反应提供了更好的候选者。此外,我们发现Co@gC 3 N 4上CO 2还原的首选产物是CO,其限速势垒为0.97 eV,而在Co 2 O 2 @gC 3 N 4上,CO 2还原为甲酸盐速率决定势垒为 0.44 eV。我们确定从COOH 生成CO 2仅发生在Co 2 O 2 @gC 3 N 4上,势垒为2.27 eV(相对较高)。为了更容易地获得上述反应的过渡态能量,我们应用机器学习方法来搜索这些量的高重要性描述符,在 C 3 N 4上负载单个过渡金属原子的情况下。有趣的是,我们的结果表明,我们感兴趣的量与基元反应产物的吸附能和归一化价电子以及金属原子的吸附能密切相关。这两组特征中的前者可以通过 DFT 直接获得,而后者的能量则被广泛列出。我们的结果为C 3 N 4负载的单金属原子上CO 2还原的催化剂和光催化剂的设计提供了指导。
更新日期:2024-04-05
中文翻译:
g-C3N4 负载的 Co 和团簇 Co2O2 光催化还原 CO2 效率:密度泛函理论和机器学习研究
从实验结果可知,gC 3 N 4负载的单原子钴是一种有效的将CO 2还原为CO的光催化剂,其光催化活性比gC 3 N 4更好。在这项工作中,我们研究了负载在石墨氮化碳(gC 3 N 4)上的钴和Co 2 O 2簇单原子的CO 2还原催化剂的性能。采用密度泛函理论加上 Hubbard (DFT + U ) 计算,我们首次详细研究了 CO 和 HCOOH 的还原机制。我们发现钴在gC 3 N 4上的沉积将gC 3 N 4的功函数降低至6.6 eV,并为还原反应提供了更好的候选者。此外,我们发现Co@gC 3 N 4上CO 2还原的首选产物是CO,其限速势垒为0.97 eV,而在Co 2 O 2 @gC 3 N 4上,CO 2还原为甲酸盐速率决定势垒为 0.44 eV。我们确定从COOH 生成CO 2仅发生在Co 2 O 2 @gC 3 N 4上,势垒为2.27 eV(相对较高)。为了更容易地获得上述反应的过渡态能量,我们应用机器学习方法来搜索这些量的高重要性描述符,在 C 3 N 4上负载单个过渡金属原子的情况下。有趣的是,我们的结果表明,我们感兴趣的量与基元反应产物的吸附能和归一化价电子以及金属原子的吸附能密切相关。这两组特征中的前者可以通过 DFT 直接获得,而后者的能量则被广泛列出。我们的结果为C 3 N 4负载的单金属原子上CO 2还原的催化剂和光催化剂的设计提供了指导。


























 京公网安备 11010802027423号
京公网安备 11010802027423号