Nature Communications ( IF 16.6 ) Pub Date : 2024-03-27 , DOI: 10.1038/s41467-024-46715-9 Gabriel Monteiro da Silva , Jennifer Y. Cui , David C. Dalgarno , George P. Lisi , Brenda M. Rubenstein
|
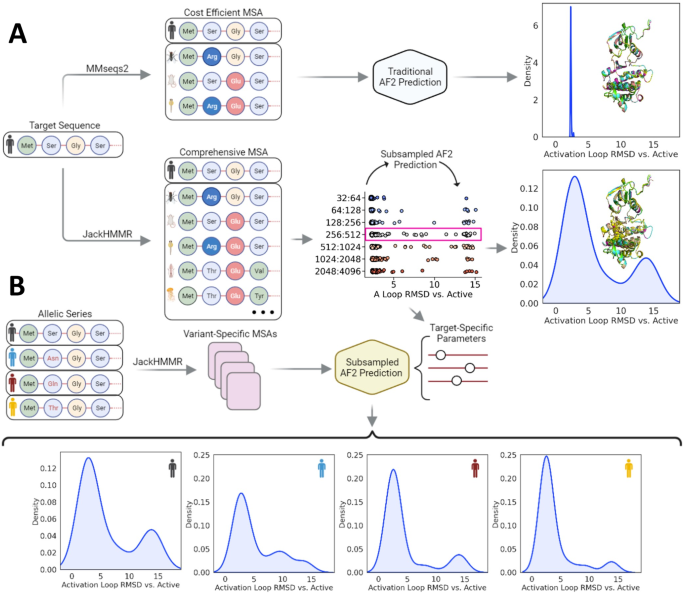
This paper presents an innovative approach for predicting the relative populations of protein conformations using AlphaFold 2, an AI-powered method that has revolutionized biology by enabling the accurate prediction of protein structures. While AlphaFold 2 has shown exceptional accuracy and speed, it is designed to predict proteins’ ground state conformations and is limited in its ability to predict conformational landscapes. Here, we demonstrate how AlphaFold 2 can directly predict the relative populations of different protein conformations by subsampling multiple sequence alignments. We tested our method against nuclear magnetic resonance experiments on two proteins with drastically different amounts of available sequence data, Abl1 kinase and the granulocyte-macrophage colony-stimulating factor, and predicted changes in their relative state populations with more than 80% accuracy. Our subsampling approach worked best when used to qualitatively predict the effects of mutations or evolution on the conformational landscape and well-populated states of proteins. It thus offers a fast and cost-effective way to predict the relative populations of protein conformations at even single-point mutation resolution, making it a useful tool for pharmacology, analysis of experimental results, and predicting evolution.
中文翻译:
使用子采样 AlphaFold2 高通量预测蛋白质构象分布
本文提出了一种使用 AlphaFold 2 预测蛋白质构象相对种群的创新方法,这是一种人工智能驱动的方法,通过准确预测蛋白质结构,彻底改变了生物学。虽然 AlphaFold 2 显示出卓越的准确性和速度,但它旨在预测蛋白质的基态构象,并且预测构象景观的能力有限。在这里,我们演示了 AlphaFold 2 如何通过对多个序列比对进行二次采样来直接预测不同蛋白质构象的相对群体。我们针对两种蛋白质(Abl1 激酶和粒细胞-巨噬细胞集落刺激因子)的可用序列数据量截然不同的核磁共振实验测试了我们的方法,并以超过 80% 的准确度预测了它们相对状态群体的变化。当用于定性预测突变或进化对蛋白质构象景观和密集状态的影响时,我们的二次采样方法效果最佳。因此,它提供了一种快速且经济高效的方法来预测蛋白质构象的相对群体,甚至是单点突变分辨率,使其成为药理学、实验结果分析和预测进化的有用工具。


























 京公网安备 11010802027423号
京公网安备 11010802027423号