Advanced Synthesis & Catalysis ( IF 5.4 ) Pub Date : 2024-03-21 , DOI: 10.1002/adsc.202301411 Janis Zakis 1 , Antonis Messinis 2 , Lutz Ackermann 3 , Tomas Smejkal 1 , Joanna Wencel-Delord 4
|
Introduction
Over the last two decades, C−H bond activation has transitioned from an academic curiosity to a practical methodology with applications in drug synthesis.1 Among the diversity of bond-forming reactions possible via C−H activation, C−H borylation has been established as a particularly attractive transformation due to the synthetic versatility of the resulting C−B bond.2 Various rhodium,3 cobalt,4 and ruthenium5 based catalytic systems have been designed. However, iridium-catalyzed C−H borylation remains the most effective and frequently used.6 The initial reports spearheaded by Hartwig, Miyaura, Ishiyama (Figure 1a), as well as Smith focused on undirected C−H borylations allowing functionalization of the most sterically accessible and/or electronically favored C−H bond.6b, 7 More recently, directed C−H borylations have emerged, in which a coordinating motif (directing group DG) governs the regioselectivity of the direct C−B coupling.6c, 8 The combination of a precisely designed DG preinstalled on the substrate with an appropriate ligand and an Ir-based precatalyst has led to complementary strategies towards ortho-, meta- and para-selective direct borylations of aromatic and heteroaromatic substrates.6c

Iridium-catalyzed ortho-directed C−H borylation.
The pioneering work of Hartwig illustrated the potential of silane DG (Figure 1b, B) in combination with bipyridine-type ligands to favor the desired ortho-borylation.9 Subsequently, multiple groups have reported ortho-directed C−H borylation using various catalytic systems including mainly phosphine ligands10 and N,N-ligands (Figure 1b, C).11 Several reports have also proposed metallacyclic intermediates as key active catalyst species.11d, 11e, 12 Despite the efficiency of such systems further improvements are desired such as, expanding of the DG scope, circumventing a need of a substrate's excess, and replacement of the air and water-sensitive Ir complex – [Ir(COD)OMe]2.13
The chemistry of metallacycles has attracted scientific curiosity for decades.14 Pioneering efforts of organometallic chemists were devoted to synthesizing such compounds via stoichiometric reactions between an organic molecule and a metal precursor,14a-14d thus providing a basis for the later development of catalytic C−H activation reactions.15 Over the last few years, various pallada-, ruthena-, and irida-cyclic species have been synthesized, and their catalytic activity has been confirmed in various direct functionalization reactions.16 Based on these observations, the use of metallacyclic species as isolable catalysts for challenging C−H activation reactions has emerged, providing not only more reactive systems but also allowing the design of novel C−H activation reactions.15d, 17
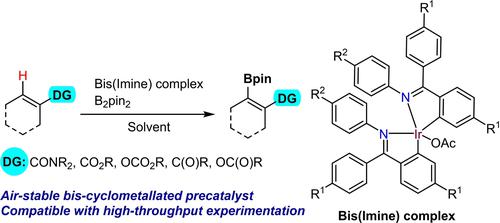
Over the years, diversity of iridium precatalysts has shown their capacity to induce the ortho-directed C−H borylation (Figure 1c, D, E).10c, 18 More recently, Chattopadhyay reported for the first time the potential of metallacyclic iridium(I) and iridium(III) precatalysts containing the 2-thienylpyridine (C,N-ligand) for C−H borylation (Figure 1c, F).19 However, the air-stable complex F requires an elevated reaction temperature (up to 120 °C in THF) and an increased B2pin2 loading to promote the desired C−H functionalization efficiently, thus rendering this catalytic system inadequate for the high-throughput experimentations (HTE). Indeed, the reaction discovery via HTE approach requires use of stable and easy to handle catalysts, to avoid experimental difficulties while operating. Therefore, intrigued by the reactivity of metallacyclic species as catalysts in C−H activation reactions, we hypothesized that the careful design of alternative C,N ligands could allow for the preparation of more catalytically active, air-stable and well-defined iridium complexes for direct borylations. Such an air-stable single component catalyst would prevent multicomponent weighing of air and water-sensitive materials and avoid solvent dependence of the catalyst preformation process. Additionally, the use of well-defined metallacyclic catalysts may aid fundamental mechanistic understanding of other in situ formed catalytic systems.
Herein we report the development of air-stable bis-cyclometallated iridium(III) precatalysts and their use in ortho-directed borylation of aromatic, heteroaromatic, acrylic C−H bonds, as well as a selected example of C(sp3)−H borylation. The precatalysts are formed from iridium acetate and bear two imine ligands, one of which gets released during the precatalyst activation.
中文翻译:
用于邻位定向 C(sp2)−H 硼化的空气稳定双环金属化铱催化剂
介绍
在过去的二十年中,C−H 键激活已经从学术好奇转变为在药物合成中应用的实用方法。1在通过 C−H 活化可能发生的成键反应的多样性中,由于所得 CB 键的合成多功能性,C−H 硼化反应已被确定为特别有吸引力的转化。2各种铑、3钴、4和钌5基催化系统已被设计。然而,铱催化的 C−H 硼化仍然是最有效和最常用的。6由 Hartwig、Miyaura、Ishiyama(图 1a)以及 Smith 牵头的初步报告重点关注无向 C−H 硼化,从而实现空间上最容易接近和/或电子最有利的 C−H 键的功能化。6b, 7最近,出现了定向 C−H 硼化,其中配位基序(定向基团 DG)控制着直接 CB 偶联的区域选择性。6c, 8预先安装在基材上的精确设计的 DG 与适当的配体和 Ir 基预催化剂的组合产生了针对芳香族和杂芳香族基材的邻位、间位和对位选择性直接硼化的互补策略。 6c
铱催化的邻位定向C−H 硼化。
Hartwig 的开创性工作说明了硅烷 DG(图 1b, B)与联吡啶型配体结合有利于所需的邻硼基化的潜力。9随后,多个研究小组报道了使用各种催化系统进行邻位 C−H 硼基化,主要包括膦配体10和N , N -配体(图 1b、C)。11一些报告还提出金属环中间体作为关键的活性催化剂种类。11d、11e、12尽管此类系统效率较高,但仍需要进一步改进,例如扩大 DG 范围、避免基材过量的需要以及更换对空气和水敏感的 Ir 络合物 – [Ir(COD)OMe ] 2.13
几十年来,金属环的化学性质一直吸引着科学界的好奇心。14有机金属化学家的开创性努力致力于通过有机分子和金属前体14a-14d之间的化学计量反应合成此类化合物,从而为后来催化 C−H 活化反应的发展奠定了基础。15在过去几年中,各种 pallada-、ruthena- 和 irida- 环状物质已被合成,并且它们的催化活性已在各种直接官能化反应中得到证实。16基于这些观察,出现了使用金属环物质作为具有挑战性的 C−H 活化反应的可分离催化剂,不仅提供了更多的反应系统,而且还允许设计新颖的 C−H 活化反应。15日、17日
多年来,铱预催化剂的多样性已显示出它们诱导邻位 C−H 硼化的能力(图 1c、D、E)。10c, 18最近,Chattopadhyay 首次报道了含有 2-噻吩基吡啶(C,N-配体)的金属环铱(I)和铱(III)预催化剂用于 C−H 硼基化的潜力(图 1c,F)。19然而,空气稳定的配合物F需要升高反应温度(在 THF 中高达 120 °C)和增加 B 2 pin 2负载量才能有效促进所需的 CH 官能化,从而使该催化系统不足以满足高反应要求。 -吞吐量实验(HTE)。事实上,通过 HTE 方法发现反应需要使用稳定且易于处理的催化剂,以避免操作时的实验困难。因此,出于对金属环物质在 C−H 活化反应中作为催化剂的反应活性的兴趣,我们假设精心设计替代的 C,N 配体可以制备更具催化活性、空气稳定性和结构明确的铱配合物,用于直接硼化。这种空气稳定的单组分催化剂将防止对空气和水敏感材料进行多组分称量,并避免催化剂预形成过程的溶剂依赖性。此外,使用明确的金属环催化剂可能有助于理解其他原位形成的催化系统的基本机理。
在此,我们报告了空气稳定的双环金属化铱(III)预催化剂的开发及其在芳香族、杂芳香族、丙烯酸C−H键的邻位硼基化中的应用,以及C( sp 3 )−H的选定示例硼酸化。预催化剂由乙酸铱形成,并带有两个亚胺配体,其中一个在预催化剂活化过程中被释放。


























 京公网安备 11010802027423号
京公网安备 11010802027423号