当前位置:
X-MOL 学术
›
Sustain. Energy Fuels
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
Lithium decorated 2D orthorhombic (o)-B2X2 monolayers for hydrogen storage: first principles calculations
Sustainable Energy & Fuels ( IF 5.6 ) Pub Date : 2024-02-28 , DOI: 10.1039/d4se00173g Ayoub Benaddi 1 , Abdelali Elomrani 1 , Mohammed Lamhani 1 , Said Oukahou 1 , Mohammad Maymoun 1 , Mohamed Yassine Fatihi 1 , Abdellatif Hasnaoui 1
Sustainable Energy & Fuels ( IF 5.6 ) Pub Date : 2024-02-28 , DOI: 10.1039/d4se00173g Ayoub Benaddi 1 , Abdelali Elomrani 1 , Mohammed Lamhani 1 , Said Oukahou 1 , Mohammad Maymoun 1 , Mohamed Yassine Fatihi 1 , Abdellatif Hasnaoui 1
Affiliation
|
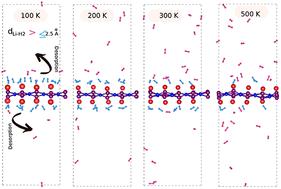
Over the last few years, scientists and researchers have shown significant interest in the search for suitable two-dimensional (2D) materials that can store hydrogen efficiently, possess high gravimetric capacity, and demonstrate excellent physisorption properties for hydrogen molecules. In this regard, we have investigated, using density functional theory (DFT) calculations, lithium decorated 2D orthorhombic (o)-B2X2 monolayers (X ≡ P or N atoms) as possible solid-state and lightweight candidate materials for hydrogen storage. Our findings reveal that o-B2P2 exhibits semi-metallic behavior, while o-B2N2 exhibits a semi-conductor state with a band gap of 0.64 eV. During the lithium decoration process, lithium adatoms exhibited strong binding energies of −3.09 and −1.98 eV for o-B2P2 and o-B2N2, respectively. These energies are significantly higher than the cohesive energy of Li (−1.63 eV), suggesting the absence of lithium-cluster formation. Furthermore, the lithium decoration process effectively enhances the adsorption of H2 molecules on both materials (e.g., 32H2@B2P2 and 24H2@B2N2), leading to high gravimetric hydrogen storage capacities of 8.18 and 9.7 wt%, respectively. With reference to an average hydrogen adsorption energy of 0.18 (for 32H2@B2P2) and 0.20 eV (24H2@B2N2), the corresponding desorption temperatures were found to be 126 and 148 K. Based on these results, it can be deduced that Li-decorated (o)-B2P2 and (o)-B2N2 hold great promise as highly effective substrates for H2 storage.
中文翻译:

用于储氢的锂装饰二维正交 (o)-B2X2 单分子层:第一原理计算
在过去的几年中,科学家和研究人员对寻找合适的二维(2D)材料表现出了浓厚的兴趣,这些材料可以有效地储存氢,具有高重力容量,并对氢分子表现出优异的物理吸附特性。在这方面,我们使用密度泛函理论(DFT)计算研究了锂装饰的2D斜方(o)-B 2 X 2单层(X == P或N原子)作为可能的固态和轻质储氢候选材料。我们的研究结果表明,oB 2 P 2表现出半金属行为,而oB 2 N 2表现出带隙为0.64 eV的半导体态。在锂装饰过程中,锂吸附原子对oB 2 P 2和oB 2 N 2分别表现出-3.09和-1.98 eV的强结合能。这些能量明显高于Li的内聚能(-1.63 eV),表明不存在锂团簇形成。此外,锂装饰过程有效增强了两种材料(例如32H 2 @B 2 P 2和24H 2 @B 2 N 2 )上H 2分子的吸附,从而实现了8.18和9.7 wt%的高重量储氢容量。 , 分别。参考平均氢吸附能为0.18(32H 2 @B 2 P 2)和0.20 eV(24H 2 @B 2 N 2),相应的解吸温度为126和148 K。基于这些结果,可以推断Li修饰的(o)-B 2 P 2和(o)-B 2 N 2作为高效H 2存储底物具有广阔的前景。
更新日期:2024-02-28
中文翻译:
用于储氢的锂装饰二维正交 (o)-B2X2 单分子层:第一原理计算
在过去的几年中,科学家和研究人员对寻找合适的二维(2D)材料表现出了浓厚的兴趣,这些材料可以有效地储存氢,具有高重力容量,并对氢分子表现出优异的物理吸附特性。在这方面,我们使用密度泛函理论(DFT)计算研究了锂装饰的2D斜方(o)-B 2 X 2单层(X == P或N原子)作为可能的固态和轻质储氢候选材料。我们的研究结果表明,oB 2 P 2表现出半金属行为,而oB 2 N 2表现出带隙为0.64 eV的半导体态。在锂装饰过程中,锂吸附原子对oB 2 P 2和oB 2 N 2分别表现出-3.09和-1.98 eV的强结合能。这些能量明显高于Li的内聚能(-1.63 eV),表明不存在锂团簇形成。此外,锂装饰过程有效增强了两种材料(例如32H 2 @B 2 P 2和24H 2 @B 2 N 2 )上H 2分子的吸附,从而实现了8.18和9.7 wt%的高重量储氢容量。 , 分别。参考平均氢吸附能为0.18(32H 2 @B 2 P 2)和0.20 eV(24H 2 @B 2 N 2),相应的解吸温度为126和148 K。基于这些结果,可以推断Li修饰的(o)-B 2 P 2和(o)-B 2 N 2作为高效H 2存储底物具有广阔的前景。


























 京公网安备 11010802027423号
京公网安备 11010802027423号