Solid State Communications ( IF 2.1 ) Pub Date : 2023-12-13 , DOI: 10.1016/j.ssc.2023.115413 Olusola G. Adeleye , Bamidele I. Adetunji , Abdulahi N. Njah , Olasunkanmi I. Olusola
|
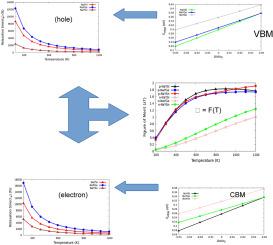
In this study, we applied Density Functional Theory (DFT) and Boltzmann Transport Theory to investigate the structural, electronic, lattice dynamic, elastic and thermometric properties of new half-Heusler compounds NaYZ (Z = Si, Ge, Sn). The structural optimization calculations revealed that the ZXY phase have the most stable state with the least energy among others. Our calculated lattice constants in are found to be 6.861, 6.899, and 7.284 for NaYZ (Z = Si, Ge, Sn). Also, the calculated elastic constants satisfied the Born-Huang criteria and were found to be mechanically and elastically stable. The Density Functional Perturbation Theory (DFPT) was used to examined the lattice dynamics of the compounds and the results shown non-existence of negative frequencies all through dispersion band structures, thus all the compounds are dynamically stable. The Seebeck coefficient of the compounds at room temperature for the p-type (n-type) are 143.79 (−48.91) V/K, 132.49 (−48.86) V/K, 154.75 (−60.24) V/k for NaYZ (Z = Si, Ge, Sn). The modified Slack's approach was used to compute lattice thermal conductivity within the experimental range and the values obtained for NaYZ (Z = Si, Ge, Sn) at room temperature are 17.265 Wm −1 K −1, 14.671 W , 10.693 W , respectively. The computations of charge carriers relaxation time ( = F(T)) as a function of temperature through deformation potential theory and the effective mass of charge carriers were used to evaluate the figure of merit (zT) for p-type and n-type of the materials. The maximum zT at 1200 K of = F(T) ( = s) are 1.76 (1.67), 1.73 (1.65), and 1.91 (1.88). These results show that = F(T) has improvement in determining the figure of merit as compared to the constant relaxation time ().
中文翻译:
新预测的半高斯勒合金 NaYZ(Z = Si、Ge、Sn)的结构、电子、弹性、晶格动力学和热电性能的第一性原理研究
在本研究中,我们应用密度泛函理论(DFT)和玻尔兹曼输运理论来研究新型半霍斯勒化合物NaYZ(Z = Si、Ge、Sn)的结构、电子、晶格动力学、弹性和测温性质。结构优化计算表明,ZXY相具有最稳定的状态和最少的能量。我们计算出的晶格常数NaYZ(Z = Si、Ge、Sn)的值分别为 6.861、6.899 和 7.284。此外,计算出的弹性常数满足 Born-Huang 标准,并且具有机械和弹性稳定性。采用密度泛函微扰理论(DFPT)研究了化合物的晶格动力学,结果表明整个色散带结构不存在负频率,因此所有化合物都是动态稳定的。p型(n型)化合物在室温下的塞贝克系数为143.79(−48.91)V/K,132.49 (−48.86)V/K,154.75 (−60.24)NaYZ(Z = Si、Ge、Sn)的 V/k。改进的 Slack 方法用于计算晶格热导率在实验范围内和NaYZ(Z = Si、Ge、Sn)在室温下获得的值为 17.265 Wm -1 K -1、 14.671 W, 10.693 瓦, 分别。载流子弛豫时间的计算( = F(T)) 通过变形势理论作为温度的函数,并使用载流子的有效质量来评估 p 型和 n 型材料的品质因数 (zT)。1200 K 时的最大 zT = F(T) ( = s) 为 1.76 (1.67)、1.73 (1.65) 和 1.91 (1.88)。这些结果表明 = F(T) 与恒定弛豫时间相比,在确定品质因数方面有所改进 ()。


























 京公网安备 11010802027423号
京公网安备 11010802027423号