npj Quantum Information ( IF 7.6 ) Pub Date : 2023-11-03 , DOI: 10.1038/s41534-023-00778-6 Taichi Kosugi , Hirofumi Nishi , Yu-ichiro Matsushita
|
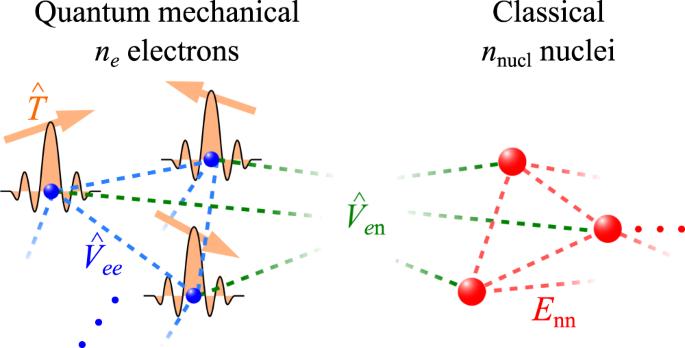
This study proposes a nonvariational scheme for geometry optimization of molecules for the first-quantized eigensolver, which is a recently proposed framework for quantum chemistry using probabilistic imaginary-time evolution (PITE). In this scheme, the nuclei in a molecule are treated as classical point charges while the electrons are treated as quantum mechanical particles. The electronic states and candidate geometries are encoded as a superposition of many-qubit states, for which a histogram created from repeated measurements gives the global minimum of the energy surface. We demonstrate that the circuit depth per step scales as \({{{\mathcal{O}}}}({n}_{{\rm {e}}}^{2}{{{\rm{poly}}}}(\log {n}_{{\rm {e}}}))\) for the electron number ne, which can be reduced to \({{{\mathcal{O}}}}({n}_{{\rm {e}}}{{{\rm{poly}}}}(\log {n}_{{\rm {e}}}))\) if extra \({{{\mathcal{O}}}}({n}_{{\rm {e}}}\log {n}_{{\rm {e}}})\) qubits are available. Moreover, resource estimation implies that the total computational time of our scheme starting from a good initial guess may exhibit overall quantum advantage in molecule size and candidate number. The proposed scheme is corroborated using numerical simulations. Additionally, a scheme adapted to variational calculations is examined that prioritizes saving circuit depths for noisy intermediate-scale quantum (NISQ) devices. A classical system composed only of charged particles is considered as a special case of the scheme. The new efficient scheme will assist in achieving scalability in practical quantum chemistry on quantum computers.
中文翻译:
在量子计算机上使用虚时间演化来详尽搜索最佳分子几何形状
这项研究提出了一种用于第一量化本征求解器的分子几何优化的非变分方案,这是最近提出的使用概率虚时演化(PITE)的量子化学框架。在该方案中,分子中的原子核被视为经典点电荷,而电子被视为量子力学粒子。电子状态和候选几何形状被编码为多量子位状态的叠加,通过重复测量创建的直方图给出了能量表面的全局最小值。我们证明每步的电路深度缩放为\({{{\mathcal{O}}}}({n}_{{\rm {e}}}^{2}{{{\rm{poly}} }}(\log {n}_{{\rm {e}}}))\)为电子数n e,可以简化为\({{{\mathcal{O}}}}({n }_{{\rm {e}}}{{{\rm{poly}}}}(\log {n}_{{\rm {e}}}))\) 如果额外 \({{ { \ mathcal{O}}}}({n}_{{\rm {e}}}\log {n}_{{\rm {e}}})\) 量子位可用。此外,资源估计意味着我们的方案从良好的初始猜测开始的总计算时间可能会在分子大小和候选数量方面表现出整体量子优势。使用数值模拟证实了所提出的方案。此外,还研究了一种适合变分计算的方案,该方案优先考虑为噪声中等规模量子(NISQ)设备节省电路深度。仅由带电粒子组成的经典系统被视为该方案的特例。新的高效方案将有助于在量子计算机上实现实用量子化学的可扩展性。


























 京公网安备 11010802027423号
京公网安备 11010802027423号