当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Multiscale Simulations of the Covalent Inhibition of the SARS-CoV-2 Main Protease: Four Compounds and Three Reaction Mechanisms
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-06-09 , DOI: 10.1021/jacs.3c02229 Bella L Grigorenko 1, 2 , Igor V Polyakov 1, 2 , Maria G Khrenova 1, 3 , Goran Giudetti 4 , Shirin Faraji 5 , Anna I Krylov 4 , Alexander V Nemukhin 1, 2
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2023-06-09 , DOI: 10.1021/jacs.3c02229 Bella L Grigorenko 1, 2 , Igor V Polyakov 1, 2 , Maria G Khrenova 1, 3 , Goran Giudetti 4 , Shirin Faraji 5 , Anna I Krylov 4 , Alexander V Nemukhin 1, 2
Affiliation
|
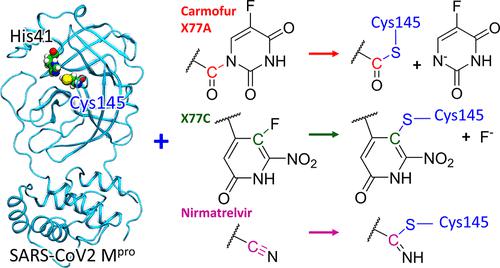
We report the results of computational modeling of the reactions of the SARS-CoV-2 main protease (MPro) with four potential covalent inhibitors. Two of them, carmofur and nirmatrelvir, have shown experimentally the ability to inhibit MPro. Two other compounds, X77A and X77C, were designed computationally in this work. They were derived from the structure of X77, a non-covalent inhibitor forming a tight surface complex with MPro. We modified the X77 structure by introducing warheads capable of reacting with the catalytic cysteine residue in the MPro active site. The reaction mechanisms of the four molecules with MPro were investigated by quantum mechanics/molecular mechanics (QM/MM) simulations. The results show that all four compounds form covalent adducts with the catalytic cysteine Cys 145 of MPro. From the chemical perspective, the reactions of these four molecules with MPro follow three distinct mechanisms. The reactions are initiated by a nucleophilic attack of the thiolate group of the deprotonated cysteine residue from the catalytic dyad Cys145–His41 of MPro. In the case of carmofur and X77A, the covalent binding of the thiolate to the ligand is accompanied by the formation of the fluoro-uracil leaving group. The reaction with X77C follows the nucleophilic aromatic substitution SNAr mechanism. The reaction of MPro with nirmatrelvir (which has a reactive nitrile group) leads to the formation of a covalent thioimidate adduct with the thiolate of the Cys145 residue in the enzyme active site. Our results contribute to the ongoing search for efficient inhibitors of the SARS-CoV-2 enzymes.
中文翻译:

SARS-CoV-2 主要蛋白酶共价抑制的多尺度模拟:四种化合物和三种反应机制
我们报告了 SARS-CoV-2 主要蛋白酶 (M Pro ) 与四种潜在共价抑制剂反应的计算模型结果。其中两种药物,卡莫氟和尼尔马韦,已通过实验显示出抑制 M Pro 的能力。本工作中通过计算设计了另外两种化合物 X77A 和 X77C。它们源自 X77 的结构,X77 是一种与 M Pro形成紧密表面复合物的非共价抑制剂。我们通过引入能够与 M Pro活性位点中的催化半胱氨酸残基反应的弹头来修改 X77 结构。通过量子力学/分子力学(QM/MM)模拟研究了四种分子与M Pro的反应机理。结果表明,所有四种化合物均与 M Pro的催化半胱氨酸 Cys 145 形成共价加合物。从化学角度来看,这四种分子与 M Pro的反应遵循三种不同的机制。该反应由 M Pro催化二元体 Cys145-His41 的去质子化半胱氨酸残基的硫醇基团的亲核攻击引发。在卡莫氟和X77A的情况下,硫醇盐与配体的共价结合伴随着氟尿嘧啶离去基团的形成。与 X77C 的反应遵循亲核芳族取代 S N Ar 机理。M Pro与 nirmatrelvir(具有反应性腈基团)的反应导致与酶活性位点的 Cys145 残基的硫醇盐形成共价硫代亚氨酸盐加合物。我们的研究结果有助于持续寻找 SARS-CoV-2 酶的有效抑制剂。
更新日期:2023-06-09
中文翻译:
SARS-CoV-2 主要蛋白酶共价抑制的多尺度模拟:四种化合物和三种反应机制
我们报告了 SARS-CoV-2 主要蛋白酶 (M Pro ) 与四种潜在共价抑制剂反应的计算模型结果。其中两种药物,卡莫氟和尼尔马韦,已通过实验显示出抑制 M Pro 的能力。本工作中通过计算设计了另外两种化合物 X77A 和 X77C。它们源自 X77 的结构,X77 是一种与 M Pro形成紧密表面复合物的非共价抑制剂。我们通过引入能够与 M Pro活性位点中的催化半胱氨酸残基反应的弹头来修改 X77 结构。通过量子力学/分子力学(QM/MM)模拟研究了四种分子与M Pro的反应机理。结果表明,所有四种化合物均与 M Pro的催化半胱氨酸 Cys 145 形成共价加合物。从化学角度来看,这四种分子与 M Pro的反应遵循三种不同的机制。该反应由 M Pro催化二元体 Cys145-His41 的去质子化半胱氨酸残基的硫醇基团的亲核攻击引发。在卡莫氟和X77A的情况下,硫醇盐与配体的共价结合伴随着氟尿嘧啶离去基团的形成。与 X77C 的反应遵循亲核芳族取代 S N Ar 机理。M Pro与 nirmatrelvir(具有反应性腈基团)的反应导致与酶活性位点的 Cys145 残基的硫醇盐形成共价硫代亚氨酸盐加合物。我们的研究结果有助于持续寻找 SARS-CoV-2 酶的有效抑制剂。










































 京公网安备 11010802027423号
京公网安备 11010802027423号