当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First principles reaction discovery: from the Schrodinger equation to experimental prediction for methane pyrolysis
Chemical Science ( IF 7.6 ) Pub Date : 2023-06-09 , DOI: 10.1039/d3sc01202f Rui Xu 1, 2 , Jan Meisner 1, 2 , Alexander M Chang 1, 2 , Keiran C Thompson 1, 2 , Todd J Martínez 1, 2
Chemical Science ( IF 7.6 ) Pub Date : 2023-06-09 , DOI: 10.1039/d3sc01202f Rui Xu 1, 2 , Jan Meisner 1, 2 , Alexander M Chang 1, 2 , Keiran C Thompson 1, 2 , Todd J Martínez 1, 2
Affiliation
|
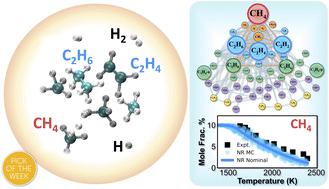
Our recent success in exploiting graphical processing units (GPUs) to accelerate quantum chemistry computations led to the development of the ab initio nanoreactor, a computational framework for automatic reaction discovery and kinetic model construction. In this work, we apply the ab initio nanoreactor to methane pyrolysis, from automatic reaction discovery to path refinement and kinetic modeling. Elementary reactions occurring during methane pyrolysis are revealed using GPU-accelerated ab initio molecular dynamics simulations. Subsequently, these reaction paths are refined at a higher level of theory with optimized reactant, product, and transition state geometries. Reaction rate coefficients are calculated by transition state theory based on the optimized reaction paths. The discovered reactions lead to a kinetic model with 53 species and 134 reactions, which is validated against experimental data and simulations using literature kinetic models. We highlight the advantage of leveraging local brute force and Monte Carlo sensitivity analysis approaches for efficient identification of important reactions. Both sensitivity approaches can further improve the accuracy of the methane pyrolysis kinetic model. The results in this work demonstrate the power of the ab initio nanoreactor framework for computationally affordable systematic reaction discovery and accurate kinetic modeling.
中文翻译:

第一原理反应发现:从薛定谔方程到甲烷热解的实验预测
我们最近在利用图形处理单元 (GPU) 加速量子化学计算方面取得了成功,从而开发了从头算纳米反应器,这是一种用于自动反应发现和动力学模型构建的计算框架。在这项工作中,我们将从头算纳米反应器应用于甲烷热解,从自动反应发现到路径细化和动力学建模。使用 GPU 加速的从头计算揭示了甲烷热解过程中发生的基本反应分子动力学模拟。随后,通过优化反应物、产物和过渡态几何形状,在更高的理论水平上完善这些反应路径。基于优化的反应路径,通过过渡态理论计算反应速率系数。发现的反应产生了包含 53 个物种和 134 个反应的动力学模型,并使用文献动力学模型根据实验数据和模拟进行了验证。我们强调利用局部强力和蒙特卡罗敏感性分析方法来有效识别重要反应的优势。两种灵敏度方法都可以进一步提高甲烷热解动力学模型的准确性。这项工作的结果证明了从头算的力量纳米反应器框架,用于计算上可负担的系统反应发现和精确的动力学建模。
更新日期:2023-06-09
中文翻译:
第一原理反应发现:从薛定谔方程到甲烷热解的实验预测
我们最近在利用图形处理单元 (GPU) 加速量子化学计算方面取得了成功,从而开发了从头算纳米反应器,这是一种用于自动反应发现和动力学模型构建的计算框架。在这项工作中,我们将从头算纳米反应器应用于甲烷热解,从自动反应发现到路径细化和动力学建模。使用 GPU 加速的从头计算揭示了甲烷热解过程中发生的基本反应分子动力学模拟。随后,通过优化反应物、产物和过渡态几何形状,在更高的理论水平上完善这些反应路径。基于优化的反应路径,通过过渡态理论计算反应速率系数。发现的反应产生了包含 53 个物种和 134 个反应的动力学模型,并使用文献动力学模型根据实验数据和模拟进行了验证。我们强调利用局部强力和蒙特卡罗敏感性分析方法来有效识别重要反应的优势。两种灵敏度方法都可以进一步提高甲烷热解动力学模型的准确性。这项工作的结果证明了从头算的力量纳米反应器框架,用于计算上可负担的系统反应发现和精确的动力学建模。










































 京公网安备 11010802027423号
京公网安备 11010802027423号