Nature Machine Intelligence ( IF 18.8 ) Pub Date : 2023-05-29 , DOI: 10.1038/s42256-023-00663-z Noah Cohen Kalafut 1, 2 , Xiang Huang 2 , Daifeng Wang 1, 2, 3
|
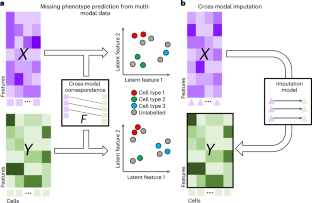
Single-cell multimodal datasets have measured various characteristics of individual cells, enabling a deep understanding of cellular and molecular mechanisms. However, multimodal data generation remains costly and challenging, and missing modalities happen frequently. Recently, machine learning approaches have been developed for data imputation but typically require fully matched multimodalities to learn common latent embeddings that potentially lack modality specificity. To address these issues, we developed an open-source machine learning model, Joint Variational Autoencoders for multimodal Imputation and Embedding (JAMIE). JAMIE takes single-cell multimodal data that can have partially matched samples across modalities. Variational autoencoders learn the latent embeddings of each modality. Then, embeddings from matched samples across modalities are aggregated to identify joint cross-modal latent embeddings before reconstruction. To perform cross-modal imputation, the latent embeddings of one modality can be used with the decoder of the other modality. For interpretability, Shapley values are used to prioritize input features for cross-modal imputation and known sample labels. We applied JAMIE to both simulation data and emerging single-cell multimodal data including gene expression, chromatin accessibility, and electrophysiology in human and mouse brains. JAMIE significantly outperforms existing state-of-the-art methods in general and prioritized multimodal features for imputation, providing potentially novel mechanistic insights at cellular resolution.
中文翻译:
用于多模态插补和嵌入的联合变分自动编码器
单细胞多模式数据集测量了单个细胞的各种特征,使人们能够深入了解细胞和分子机制。然而,多模式数据生成仍然成本高昂且具有挑战性,并且缺失模式的情况经常发生。最近,已经开发了用于数据插补的机器学习方法,但通常需要完全匹配的多模态来学习可能缺乏模态特异性的常见潜在嵌入。为了解决这些问题,我们开发了一种开源机器学习模型,即用于多模态插补和嵌入的联合变分自编码器 (JAMIE)。 JAMIE 采用单细胞多模式数据,这些数据可以跨模式部分匹配样本。变分自动编码器学习每种模态的潜在嵌入。然后,聚合来自跨模态的匹配样本的嵌入,以在重建之前识别联合跨模态潜在嵌入。为了执行跨模态插补,一种模态的潜在嵌入可以与另一种模态的解码器一起使用。为了可解释性,Shapley 值用于对跨模式插补和已知样本标签的输入特征进行优先级排序。我们将 JAMIE 应用于模拟数据和新兴的单细胞多模态数据,包括人类和小鼠大脑中的基因表达、染色质可及性以及电生理学。 JAMIE 总体上显着优于现有的最先进方法,并优先考虑用于插补的多模态特征,在细胞分辨率下提供潜在的新颖机制见解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号