当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Extrapolating Energetics on Clusters and Single-Crystal Surfaces to Nanoparticles by Machine-Learning Scheme
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2017-11-17 00:00:00 , DOI: 10.1021/acs.jpcc.7b08686 Ryosuke Jinnouchi 1 , Hirohito Hirata 2 , Ryoji Asahi 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2017-11-17 00:00:00 , DOI: 10.1021/acs.jpcc.7b08686 Ryosuke Jinnouchi 1 , Hirohito Hirata 2 , Ryoji Asahi 1
Affiliation
|
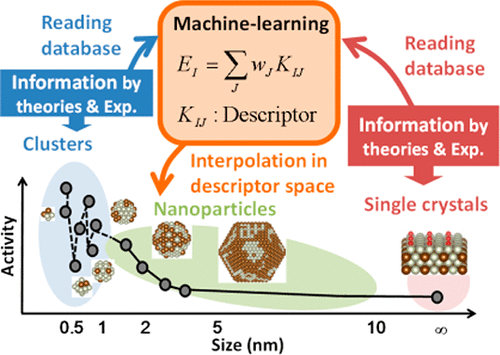
A Bayesian linear regression scheme using a local structural similarity kernel as a descriptor is used to predict the energetics of atoms and molecules on nanoparticles. Examination of the binding energies of N, O, and NO with RhAu alloy single-crystal surfaces and particles indicates that regression models predict the binding energies on nanoparticles having diameters greater than 1.5 nm within an error range of 100–150 meV when the DFT data on single-crystal surfaces are used for training. By contrast, when the DFT data on small clusters are used for training, the regression models produce an error range of 200–400 meV. Kinetic analyses using the predicted energetics of the direct decomposition of NO on RhAu nanoparticles indicate that catalytic activity increases with a decrease in the particle diameter to 2.0 nm, whereas the activity drops when the diameter decreases to 1.5 nm. Detailed examinations of the free energy diagrams and the structures of active sites indicate that the drop in catalytic activity derives from the disappearance of active alloyed corner sites on the small nanoparticles as a result of Au segregation at the corners of narrow facets.
中文翻译:

通过机器学习方案将团簇和单晶表面的能量外推到纳米粒子
使用局部结构相似性核作为描述符的贝叶斯线性回归方案可用于预测纳米粒子上原子和分子的能量。用RhAu合金单晶表面和颗粒检测N,O和NO的结合能表明,当DFT数据显示时,回归模型可预测直径大于1.5 nm的纳米颗粒的结合能在100–150 meV的误差范围内在单晶表面上用于训练。相反,当使用小簇上的DFT数据进行训练时,回归模型产生的误差范围为200–400 meV。使用Rh纳米颗粒上NO的直接分解的预测能量学进行的动力学分析表明,催化活性随粒径减小至2.0 nm而增加,而当直径减小到1.5 nm时,活性降低。对自由能图和活性位点结构的详细检查表明,催化活性的下降源于小纳米颗粒上活性合金角位点的消失,这是由于窄小面角处的Au偏析所致。
更新日期:2017-11-20
中文翻译:
通过机器学习方案将团簇和单晶表面的能量外推到纳米粒子
使用局部结构相似性核作为描述符的贝叶斯线性回归方案可用于预测纳米粒子上原子和分子的能量。用RhAu合金单晶表面和颗粒检测N,O和NO的结合能表明,当DFT数据显示时,回归模型可预测直径大于1.5 nm的纳米颗粒的结合能在100–150 meV的误差范围内在单晶表面上用于训练。相反,当使用小簇上的DFT数据进行训练时,回归模型产生的误差范围为200–400 meV。使用Rh纳米颗粒上NO的直接分解的预测能量学进行的动力学分析表明,催化活性随粒径减小至2.0 nm而增加,而当直径减小到1.5 nm时,活性降低。对自由能图和活性位点结构的详细检查表明,催化活性的下降源于小纳米颗粒上活性合金角位点的消失,这是由于窄小面角处的Au偏析所致。










































 京公网安备 11010802027423号
京公网安备 11010802027423号