Journal of Power Sources ( IF 8.1 ) Pub Date : 2017-11-08 , DOI: 10.1016/j.jpowsour.2017.10.081 Chanbum Park , Matej Kanduč , Richard Chudoba , Arne Ronneburg , Sebastian Risse , Matthias Ballauff , Joachim Dzubiella
|
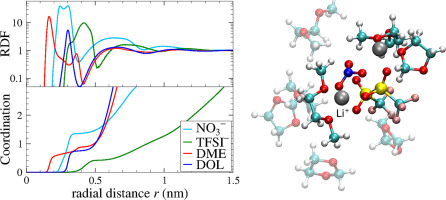
The performance of modern lithium-sulfur (Li/S) battery systems critically depends on the electrolyte and solvent compositions. For fundamental molecular insights and rational guidance of experimental developments, efficient and sufficiently accurate molecular simulations are thus in urgent need. Here, we construct a molecular dynamics (MD) computer simulation model of representative state-of-the art electrolyte–solvent systems for Li/S batteries constituted by lithium-bis(trifluoromethane)sulfonimide () and electrolytes in mixtures of the organic solvents 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL). We benchmark and verify our simulations by comparing structural and dynamic features with various available experimental reference systems and demonstrate their applicability for a wide range of electrolyte–solvent compositions. For the state-of-the-art battery solvent, we finally calculate and discuss the detailed composition of the first lithium solvation shell, the temperature dependence of lithium diffusion, as well as the electrolyte conductivities and lithium transference numbers. Our model will serve as a basis for efficient future predictions of electrolyte structure and transport in complex electrode confinements for the optimization of modern Li/S batteries (and related devices).
中文翻译:
锂硫电池溶剂中电解质结构和动力学的分子模拟
现代锂硫(Li / S)电池系统的性能关键取决于电解质和溶剂的组成。为了获得基本的分子见解和实验发展的合理指导,因此迫切需要高效且足够准确的分子模拟。在这里,我们构建了一个分子动力学(MD)计算机模拟模型,该模型代表了由锂-双(三氟甲烷)磺酰亚胺锂() 和 有机溶剂1,2-二甲氧基乙烷(DME)和1,3-二氧戊环(DOL)的混合物中的电解质。我们通过将结构和动态特征与各种可用的实验参考系统进行比较来对模拟进行基准测试和验证,并证明它们在各种电解质-溶剂组合物中的适用性。对于最新的电池溶剂,我们最终计算并讨论了第一个锂溶剂化壳的详细组成,锂扩散的温度依赖性以及电解质的电导率和锂转移数。我们的模型将作为未来有效地预测复杂锂离子电池中电解质结构和传输的基础,以优化现代Li / S电池(及相关设备)。










































 京公网安备 11010802027423号
京公网安备 11010802027423号