当前位置:
X-MOL 学术
›
J. Proteome Res.
›
论文详情
Our official English website, www.x-mol.net, welcomes your feedback! (Note: you will need to create a separate account there.)
ADAP-GC 3.2: Graphical Software Tool for Efficient Spectral Deconvolution of Gas Chromatography–High-Resolution Mass Spectrometry Metabolomics Data
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2017-11-07 00:00:00 , DOI: 10.1021/acs.jproteome.7b00633 Aleksandr Smirnov 1 , Wei Jia 2 , Douglas I. Walker 3 , Dean P. Jones 3 , Xiuxia Du 1
Journal of Proteome Research ( IF 4.4 ) Pub Date : 2017-11-07 00:00:00 , DOI: 10.1021/acs.jproteome.7b00633 Aleksandr Smirnov 1 , Wei Jia 2 , Douglas I. Walker 3 , Dean P. Jones 3 , Xiuxia Du 1
Affiliation
|
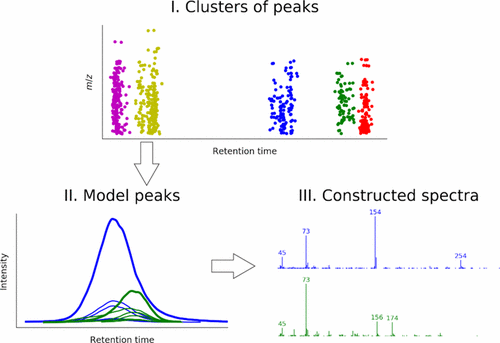
ADAP-GC is an automated computational workflow for extracting metabolite information from raw, untargeted gas chromatography–mass spectrometry metabolomics data. Deconvolution of coeluting analytes is a critical step in the workflow, and the underlying algorithm is able to extract fragmentation mass spectra of coeluting analytes with high accuracy. However, its latest version ADAP-GC 3.0 was not user-friendly. To make ADAP-GC easier to use, we have developed ADAP-GC 3.2 and describe here the improvements on three aspects. First, all of the algorithms in ADAP-GC 3.0 written in R have been replaced by their analogues in Java and incorporated into MZmine 2 to make the workflow user-friendly. Second, the clustering algorithm DBSCAN has replaced the original hierarchical clustering to allow faster spectral deconvolution. Finally, algorithms originally developed for constructing extracted ion chromatograms (EICs) and detecting EIC peaks from LC–MS data are incorporated into the ADAP-GC workflow, allowing the latter to process high mass resolution data. Performance of ADAP-GC 3.2 has been evaluated using unit mass resolution data from standard-mixture and urine samples. The identification and quantitation results were compared with those produced by ADAP-GC 3.0, AMDIS, AnalyzerPro, and ChromaTOF. Identification results for high mass resolution data derived from standard-mixture samples are presented as well.
中文翻译:

ADAP-GC 3.2:图形软件工具,用于气相色谱的高效光谱解卷积-高分辨率质谱代谢组学数据
ADAP-GC是一种自动计算工作流程,用于从原始的,无目标的气相色谱-质谱分析代谢组学数据中提取代谢物信息。共洗脱分析物的去卷积是工作流程中的关键步骤,基础算法能够以高精度提取共洗脱分析物的碎片质谱。但是,其最新版本的ADAP-GC 3.0并不友好。为了使ADAP-GC易于使用,我们开发了ADAP-GC 3.2并在此描述了三个方面的改进。首先,用R编写的ADAP-GC 3.0中的所有算法已被其Java中的类似物取代,并已合并到MZmine 2中,以使工作流变得用户友好。其次,聚类算法DBSCAN取代了原始的分层聚类,以实现更快的频谱反卷积。最后,最初为构建提取的离子色谱图(EIC)和从LC-MS数据检测EIC峰而开发的算法已被整合到ADAP-GC工作流程中,从而使后者能够处理高质量的数据。已使用标准混合物和尿液样品的单位质量分辨率数据评估了ADAP-GC 3.2的性能。鉴定和定量结果与ADAP-GC 3.0,AMDIS,AnalyzerPro和ChromaTOF产生的结果进行了比较。还显示了从标准混合物样品中获得的高质量分离度数据的鉴定结果。使用标准混合物和尿液样品的单位质量分辨率数据对图2进行了评估。鉴定和定量结果与ADAP-GC 3.0,AMDIS,AnalyzerPro和ChromaTOF产生的结果进行了比较。还显示了从标准混合物样品中获得的高质量分离度数据的鉴定结果。使用标准混合物和尿液样品的单位质量分辨率数据对图2进行了评估。鉴定和定量结果与ADAP-GC 3.0,AMDIS,AnalyzerPro和ChromaTOF产生的结果进行了比较。还显示了从标准混合物样品中获得的高质量分离度数据的鉴定结果。
更新日期:2017-11-08
中文翻译:
ADAP-GC 3.2:图形软件工具,用于气相色谱的高效光谱解卷积-高分辨率质谱代谢组学数据
ADAP-GC是一种自动计算工作流程,用于从原始的,无目标的气相色谱-质谱分析代谢组学数据中提取代谢物信息。共洗脱分析物的去卷积是工作流程中的关键步骤,基础算法能够以高精度提取共洗脱分析物的碎片质谱。但是,其最新版本的ADAP-GC 3.0并不友好。为了使ADAP-GC易于使用,我们开发了ADAP-GC 3.2并在此描述了三个方面的改进。首先,用R编写的ADAP-GC 3.0中的所有算法已被其Java中的类似物取代,并已合并到MZmine 2中,以使工作流变得用户友好。其次,聚类算法DBSCAN取代了原始的分层聚类,以实现更快的频谱反卷积。最后,最初为构建提取的离子色谱图(EIC)和从LC-MS数据检测EIC峰而开发的算法已被整合到ADAP-GC工作流程中,从而使后者能够处理高质量的数据。已使用标准混合物和尿液样品的单位质量分辨率数据评估了ADAP-GC 3.2的性能。鉴定和定量结果与ADAP-GC 3.0,AMDIS,AnalyzerPro和ChromaTOF产生的结果进行了比较。还显示了从标准混合物样品中获得的高质量分离度数据的鉴定结果。使用标准混合物和尿液样品的单位质量分辨率数据对图2进行了评估。鉴定和定量结果与ADAP-GC 3.0,AMDIS,AnalyzerPro和ChromaTOF产生的结果进行了比较。还显示了从标准混合物样品中获得的高质量分离度数据的鉴定结果。使用标准混合物和尿液样品的单位质量分辨率数据对图2进行了评估。鉴定和定量结果与ADAP-GC 3.0,AMDIS,AnalyzerPro和ChromaTOF产生的结果进行了比较。还显示了从标准混合物样品中获得的高质量分离度数据的鉴定结果。


























 京公网安备 11010802027423号
京公网安备 11010802027423号