当前位置:
X-MOL 学术
›
Chem. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Bias-dependent local structure of water molecules at a metallic interface
Chemical Science ( IF 7.6 ) Pub Date : 2017-10-11 00:00:00 , DOI: 10.1039/c7sc02208e Luana S. Pedroza 1, 2, 3, 4, 5 , Pedro Brandimarte 6, 7, 7, 8, 9 , Alexandre Reily Rocha 2, 3, 4 , M.-V. Fernández-Serra 10, 11, 12, 13, 14
Chemical Science ( IF 7.6 ) Pub Date : 2017-10-11 00:00:00 , DOI: 10.1039/c7sc02208e Luana S. Pedroza 1, 2, 3, 4, 5 , Pedro Brandimarte 6, 7, 7, 8, 9 , Alexandre Reily Rocha 2, 3, 4 , M.-V. Fernández-Serra 10, 11, 12, 13, 14
Affiliation
|
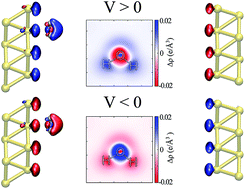
Understanding the local structure of water at the interfaces of metallic electrodes is a key issue in aqueous-based electrochemistry. Nevertheless a realistic simulation of such a setup is challenging, particularly when the electrodes are maintained at different potentials. To correctly compute the effect of an external bias potential applied to truly semi-infinite surfaces, we combine Density Functional Theory (DFT) and Non-Equilibrium Green’s Function (NEGF) methods. This framework allows for the out-of-equilibrium calculation of forces and dynamics, and directly correlates to the chemical potential of the electrodes, which is introduced experimentally. In this work, we apply this methodology to study the electronic properties and atomic forces of a water molecule at the interface of a gold surface. We find that the water molecule tends to align its dipole moment with the electric field, and it is either repelled or attracted to the metal depending on the sign and magnitude of the applied bias, in an asymmetric fashion.
中文翻译:

金属界面上水分子的偏倚局部结构
了解水在金属电极界面处的局部结构是水基电化学中的关键问题。然而,这种设置的现实模拟是具有挑战性的,特别是当电极保持在不同电位时。为了正确计算施加在真正的半无限表面上的外部偏置电势的影响,我们结合了密度泛函理论(DFT)和非平衡格林函数(NEGF)方法。该框架允许力和动力学的不平衡计算,并且与通过实验引入的电极的化学势直接相关。在这项工作中,我们将这种方法应用于研究金表面界面上水分子的电子性质和原子力。
更新日期:2017-11-03
中文翻译:
金属界面上水分子的偏倚局部结构
了解水在金属电极界面处的局部结构是水基电化学中的关键问题。然而,这种设置的现实模拟是具有挑战性的,特别是当电极保持在不同电位时。为了正确计算施加在真正的半无限表面上的外部偏置电势的影响,我们结合了密度泛函理论(DFT)和非平衡格林函数(NEGF)方法。该框架允许力和动力学的不平衡计算,并且与通过实验引入的电极的化学势直接相关。在这项工作中,我们将这种方法应用于研究金表面界面上水分子的电子性质和原子力。










































 京公网安备 11010802027423号
京公网安备 11010802027423号