当前位置:
X-MOL 学术
›
J. Nat. Prod.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Structural Sequencing of Oligopeptides Aided by 1H Iterative Full-Spin Analysis
Journal of Natural Products ( IF 3.3 ) Pub Date : 2017-10-16 00:00:00 , DOI: 10.1021/acs.jnatprod.7b00207 Wei Gao 1, 2 , James B. McAlpine 1, 2 , Mary P. Choules 1, 2 , José G. Napolitano 1, 3 , David C. Lankin 1, 3 , Charlotte Simmler 1, 3 , Ngoc Anh Ho 4 , Hanki Lee 4 , Joo-Won Suh 4, 5 , Ian W. Burton 6 , Sanghyun Cho 2 , Scott G. Franzblau 2 , Shao-Nong Chen 1, 2, 3 , Guido F. Pauli 1, 2, 3
Journal of Natural Products ( IF 3.3 ) Pub Date : 2017-10-16 00:00:00 , DOI: 10.1021/acs.jnatprod.7b00207 Wei Gao 1, 2 , James B. McAlpine 1, 2 , Mary P. Choules 1, 2 , José G. Napolitano 1, 3 , David C. Lankin 1, 3 , Charlotte Simmler 1, 3 , Ngoc Anh Ho 4 , Hanki Lee 4 , Joo-Won Suh 4, 5 , Ian W. Burton 6 , Sanghyun Cho 2 , Scott G. Franzblau 2 , Shao-Nong Chen 1, 2, 3 , Guido F. Pauli 1, 2, 3
Affiliation
|
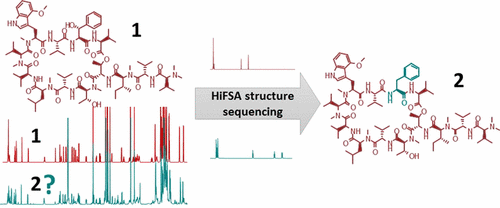
This report describes an approach using 1H NMR iterative full-spin analysis (HiFSA) to extract definitive structural information on unknown peptides from 1D 1H NMR data. By comparing the experimental data and HiFSA fingerprint of a known analogue, it is possible to isolate the characteristic 1H subspectrum of the different amino acids and, thus, elucidate the structure of the peptide. To illustrate this methodology, a comprehensive analysis of five new anti-Mycobacterium tuberculosis peptides (2–6), all analogues of ecumicin (1), was carried out. The method was validated by demonstrating congruence of the HiFSA-based structures with all available data, including MS and 2D NMR. The highly reproducible HiFSA fingerprints of the new ∼1600 amu peptides were generated in this process. Besides oligo-peptides, the HiFSA sequencing approach could be extended to all oligomeric compounds consisting of chains of monomers lacking H–H spin–spin coupling across the moieties. HiFSA sequencing is capable of differentiating complex oligomers that exhibit minor structural differences such as shifted hydoxyl or methyl groups. Because it employs the basic and most sensitive 1D 1H NMR experiment, HiFSA sequencing enables the exploration of peptide analogues up to at least 2000 amu, even with basic contemporary spectrometers and when only sub-milligram amounts of isolates are available.
中文翻译:

1 H迭代全自旋分析辅助寡肽的结构测序。
本报告介绍了一种使用1 H NMR迭代全旋波分析(HiFSA)从1D 1 H NMR数据中提取未知肽的确定结构信息的方法。通过比较实验数据和已知类似物的HiFSA指纹,可以分离出不同氨基酸的特征性1 H亚谱,从而阐明肽的结构。为了说明这种方法,对5种新的抗结核分枝杆菌肽(2 – 6),所有的ecumicin类似物(1),已执行。通过证明基于HiFSA的结构与所有可用数据(包括MS和2D NMR)的一致性,验证了该方法。在此过程中产生了新的〜1600 amu肽的高度可复制的HiFSA指纹。除寡肽外,HiFSA测序方法还可以扩展到所有寡聚化合物,这些寡聚化合物由单体链组成,这些单体链之间缺乏H–H自旋-自旋耦合。HiFSA测序能够区分复杂的低聚物,这些低聚物表现出较小的结构差异,例如移位的羟基或甲基。因为它采用了基本且最敏感的1D 1在1 H NMR实验中,即使使用基本的现代光谱仪,并且只有亚毫克量的分离物,HiFSA测序也可以探索至少2000 amu的肽类似物。
更新日期:2017-10-16
中文翻译:
1 H迭代全自旋分析辅助寡肽的结构测序。
本报告介绍了一种使用1 H NMR迭代全旋波分析(HiFSA)从1D 1 H NMR数据中提取未知肽的确定结构信息的方法。通过比较实验数据和已知类似物的HiFSA指纹,可以分离出不同氨基酸的特征性1 H亚谱,从而阐明肽的结构。为了说明这种方法,对5种新的抗结核分枝杆菌肽(2 – 6),所有的ecumicin类似物(1),已执行。通过证明基于HiFSA的结构与所有可用数据(包括MS和2D NMR)的一致性,验证了该方法。在此过程中产生了新的〜1600 amu肽的高度可复制的HiFSA指纹。除寡肽外,HiFSA测序方法还可以扩展到所有寡聚化合物,这些寡聚化合物由单体链组成,这些单体链之间缺乏H–H自旋-自旋耦合。HiFSA测序能够区分复杂的低聚物,这些低聚物表现出较小的结构差异,例如移位的羟基或甲基。因为它采用了基本且最敏感的1D 1在1 H NMR实验中,即使使用基本的现代光谱仪,并且只有亚毫克量的分离物,HiFSA测序也可以探索至少2000 amu的肽类似物。










































 京公网安备 11010802027423号
京公网安备 11010802027423号