当前位置:
X-MOL 学术
›
Phys. Chem. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical study on geometric, electronic and catalytic performances of Fe dopant pairs in graphene
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2017-09-08 00:00:00 , DOI: 10.1039/c7cp05683d Yanan Tang 1, 2, 3, 4, 5 , Huadou Chai 1, 2, 3, 4, 5 , Weiguang Chen 1, 2, 3, 4, 5 , Xiao Cui 1, 2, 3, 4, 5 , Yaqiang Ma 5, 6, 7, 8 , Mingyu Zhao 5, 6, 7, 8 , Xianqi Dai 1, 2, 3, 4, 5
Physical Chemistry Chemical Physics ( IF 2.9 ) Pub Date : 2017-09-08 00:00:00 , DOI: 10.1039/c7cp05683d Yanan Tang 1, 2, 3, 4, 5 , Huadou Chai 1, 2, 3, 4, 5 , Weiguang Chen 1, 2, 3, 4, 5 , Xiao Cui 1, 2, 3, 4, 5 , Yaqiang Ma 5, 6, 7, 8 , Mingyu Zhao 5, 6, 7, 8 , Xianqi Dai 1, 2, 3, 4, 5
Affiliation
|
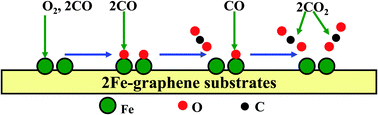
The formation geometries, electronic structures and catalytic properties of monovacancy and divacancy graphene sheets with two embedded Fe dopants (2Fe-MG and 2Fe-DG) have been systematically investigated using the first-principles calculations. It was found that the configuration of 2Fe-DG is slightly more stable than that of 2Fe-MG sheets and the two doped Fe atoms in graphene (2Fe-graphene) as active sites could regulate the stability of gas molecules. In addition, the adsorption of O2 and CO molecules could modulate the electronic and magnetic properties of 2Fe-graphene systems. Moreover, the adsorption behaviors of reactants could determine the reaction pathway and energy barrier of the catalytic oxidation of CO. On the 2Fe-graphene substrates, the adsorptive decomposition of O2 molecules (<0.20 eV) and the subsequent Eley–Rideal (ER) reaction (2Oads + 2CO → CO2) (<0.60 eV) have low energy barriers. In comparison, the CO3 complex is quite stable and its formation needs to overcome a higher energy barrier (>0.90 eV). Hence, the dissociation of O2 as an initial step is an energetically more favored process. These results provide valuable guidance for the design of functionalized graphene-based devices.
中文翻译:

石墨烯中Fe掺杂对的几何,电子和催化性能的理论研究
使用第一性原理计算系统地研究了具有两种嵌入的Fe掺杂剂(2Fe-MG和2Fe-DG)的单空位和双空位石墨烯片的形成几何形状,电子结构和催化性能。发现2Fe-DG的构型比2Fe-MG片的构型稍微稳定一些,并且石墨烯中的两个掺杂的Fe原子(2Fe-石墨烯)作为活性位点可以调节气体分子的稳定性。另外,O 2和CO分子的吸附可以调节2Fe-石墨烯体系的电子和磁性。此外,反应物的吸附行为可以决定CO催化氧化的反应途径和能垒。在2Fe-石墨烯基体上,O 2的吸附分解分子(<0.20 eV)和随后的Eley-Rideal(ER)反应(2O ads + 2CO→CO 2)(<0.60 eV)具有较低的能垒。相比之下,CO 3络合物非常稳定,其形成需要克服更高的能垒(> 0.90 eV)。因此,作为初始步骤,O 2的解离在能量上是更有利的过程。这些结果为功能化的基于石墨烯的器件的设计提供了有价值的指导。
更新日期:2017-09-22
中文翻译:
石墨烯中Fe掺杂对的几何,电子和催化性能的理论研究
使用第一性原理计算系统地研究了具有两种嵌入的Fe掺杂剂(2Fe-MG和2Fe-DG)的单空位和双空位石墨烯片的形成几何形状,电子结构和催化性能。发现2Fe-DG的构型比2Fe-MG片的构型稍微稳定一些,并且石墨烯中的两个掺杂的Fe原子(2Fe-石墨烯)作为活性位点可以调节气体分子的稳定性。另外,O 2和CO分子的吸附可以调节2Fe-石墨烯体系的电子和磁性。此外,反应物的吸附行为可以决定CO催化氧化的反应途径和能垒。在2Fe-石墨烯基体上,O 2的吸附分解分子(<0.20 eV)和随后的Eley-Rideal(ER)反应(2O ads + 2CO→CO 2)(<0.60 eV)具有较低的能垒。相比之下,CO 3络合物非常稳定,其形成需要克服更高的能垒(> 0.90 eV)。因此,作为初始步骤,O 2的解离在能量上是更有利的过程。这些结果为功能化的基于石墨烯的器件的设计提供了有价值的指导。










































 京公网安备 11010802027423号
京公网安备 11010802027423号