当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Proton Capture Dynamics in Quinoline Photobases: Substituent Effect and Involvement of Triplet States
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-09-19 00:00:00 , DOI: 10.1021/acs.jpca.7b04512 Eric William Driscoll 1 , Jonathan Ryan Hunt 1 , Jahan M. Dawlaty 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2017-09-19 00:00:00 , DOI: 10.1021/acs.jpca.7b04512 Eric William Driscoll 1 , Jonathan Ryan Hunt 1 , Jahan M. Dawlaty 1
Affiliation
|
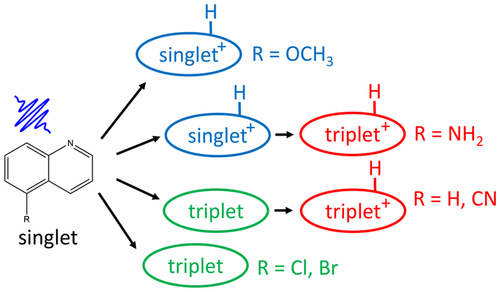
Converting light into chemical energy often occurs through redox reactions that require transfer of several electrons and protons. Using light to control proton transfer has the potential for driving otherwise unfavorable protonation reactions or producing transient pH changes. Photoacids and photobases are fundamental functional elements that could serve this purpose. Previously, we have reported the thermodynamic drive for proton removal in a series of quinoline photobases using Forster cycle analysis of the singlet states. Because the existence of thermodynamic drive does not imply that the molecules can indeed capture protons in the excited state, in this work we report the kinetics of proton removal from water by 5-R-quinolines, R = {NH2, OCH3, H, Cl, Br, CN}, using ultrafast transient absorption spectroscopy. We found that the time constants and mechanisms of proton capture from water are highly sensitive to the substituent. In some cases, proton transfer occurs within the singlet manifold, whereas in some others intersystem crossing competes with this process. We have evidence that the triplet states are also capable of proton capture in two of the compounds. This renders the excited state proton transfer process more complicated than can be captured by the linear free energy relationships inferred from the energetics of the singlet states. We have measured proton capture times in this family to be in the range of several tens of picoseconds with no discernible trend with respect to the Hammett parameter of the substituents. This wide range of mechanisms is attributed to the high density of excited electronic states in the singlet and triplet manifolds. The ordering between these states is expected to change by substituent, solvent, and hydrogen bonding, thus making the rate of intersystem crossing and proton transfer very sensitive to these parameters. These results are necessary fundamental steps to assess the capabilities of photobases in prospective applications such as photomediated proton removal in redox reactions, steady state optical regulation of local pH, and pOH jump kinetics experiments.
中文翻译:

喹啉碱的质子俘获动力学:取代效应和三重态的介入。
将光转换成化学能通常是通过需要几个电子和质子转移的氧化还原反应发生的。使用光控制质子转移有可能驱动原本不利的质子化反应或产生短暂的pH变化。光酸和光碱是可以用于此目的的基本功能元素。以前,我们已经报道了使用单峰态的Forster循环分析在一系列喹啉光碱中进行质子去除的热力学驱动力。因为热力学驱动的存在并不意味着分子确实可以在激发态下捕获质子,所以在这项工作中,我们报告了5-R-喹啉从水中去除质子的动力学,R = {NH 2,OCH 3,H,Cl,Br,CN},使用超快瞬态吸收光谱法。我们发现时间常数和质子从水中捕获的机理对取代基高度敏感。在某些情况下,质子传递发生在单线态歧管内,而在另一些情况下,系统间的交叉与此过程竞争。我们有证据表明三重态也能够在两种化合物中捕获质子。这使得激发态质子转移过程比从单重态态的能量学推断出的线性自由能关系所捕获的过程更为复杂。我们测得该族中的质子捕获时间在几十皮秒的范围内,相对于取代基的哈米特参数没有明显的趋势。这种广泛的机制归因于单重态和三重态歧管中激发电子态的高密度。预期这些状态之间的顺序会因取代基,溶剂和氢键而改变,因此使系统间交叉和质子转移的速率对这些参数非常敏感。这些结果是评估光基础在预期应用中的能力所必需的基本步骤,例如在氧化还原反应中光介导的质子去除,局部pH的稳态光学调节以及pOH跳跃动力学实验。因此,使系统间交叉和质子转移的速率对这些参数非常敏感。这些结果是评估光基础在预期应用中的能力所必需的基本步骤,例如在氧化还原反应中光介导的质子去除,局部pH的稳态光学调节以及pOH跳跃动力学实验。因此,使系统间交叉和质子转移的速率对这些参数非常敏感。这些结果是评估光基础在预期应用中的能力所必需的基本步骤,例如在氧化还原反应中光介导的质子去除,局部pH的稳态光学调节以及pOH跳跃动力学实验。
更新日期:2017-09-20
中文翻译:
喹啉碱的质子俘获动力学:取代效应和三重态的介入。
将光转换成化学能通常是通过需要几个电子和质子转移的氧化还原反应发生的。使用光控制质子转移有可能驱动原本不利的质子化反应或产生短暂的pH变化。光酸和光碱是可以用于此目的的基本功能元素。以前,我们已经报道了使用单峰态的Forster循环分析在一系列喹啉光碱中进行质子去除的热力学驱动力。因为热力学驱动的存在并不意味着分子确实可以在激发态下捕获质子,所以在这项工作中,我们报告了5-R-喹啉从水中去除质子的动力学,R = {NH 2,OCH 3,H,Cl,Br,CN},使用超快瞬态吸收光谱法。我们发现时间常数和质子从水中捕获的机理对取代基高度敏感。在某些情况下,质子传递发生在单线态歧管内,而在另一些情况下,系统间的交叉与此过程竞争。我们有证据表明三重态也能够在两种化合物中捕获质子。这使得激发态质子转移过程比从单重态态的能量学推断出的线性自由能关系所捕获的过程更为复杂。我们测得该族中的质子捕获时间在几十皮秒的范围内,相对于取代基的哈米特参数没有明显的趋势。这种广泛的机制归因于单重态和三重态歧管中激发电子态的高密度。预期这些状态之间的顺序会因取代基,溶剂和氢键而改变,因此使系统间交叉和质子转移的速率对这些参数非常敏感。这些结果是评估光基础在预期应用中的能力所必需的基本步骤,例如在氧化还原反应中光介导的质子去除,局部pH的稳态光学调节以及pOH跳跃动力学实验。因此,使系统间交叉和质子转移的速率对这些参数非常敏感。这些结果是评估光基础在预期应用中的能力所必需的基本步骤,例如在氧化还原反应中光介导的质子去除,局部pH的稳态光学调节以及pOH跳跃动力学实验。因此,使系统间交叉和质子转移的速率对这些参数非常敏感。这些结果是评估光基础在预期应用中的能力所必需的基本步骤,例如在氧化还原反应中光介导的质子去除,局部pH的稳态光学调节以及pOH跳跃动力学实验。










































 京公网安备 11010802027423号
京公网安备 11010802027423号