Polymer ( IF 4.1 ) Pub Date : 2017-09-18 , DOI: 10.1016/j.polymer.2017.09.038 Jacob R. Gissinger , Benjamin D. Jensen , Kristopher E. Wise
|
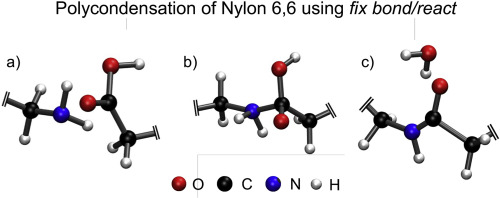
An algorithm capable of incorporating multi-step reaction mechanisms into atomistic molecular dynamics simulations using traditional fixed valence force fields is proposed and implemented within the framework of LAMMPS (Large-scale Atomic Molecular Massively Parallel Simulator). This extension, referred to as fix bond/react, enables bonding topology modifications during a running MD simulation using pre- and post-reaction bonding templates to carry out a pre-specified reaction. Candidate reactants are first identified by interatomic separation, followed by the application of a generalized topology matching algorithm to confirm they match the pre-reaction template. This is followed by a topology conversion to match the post-reaction template and a dynamic relaxation to minimize high energy configurations. Two case studies, the condensation polymerization of nylon 6,6 and the formation of a highly-crosslinked epoxy, are simulated to demonstrate the robustness, stability, and speed of the algorithm. Improvements which could increase its utility are discussed.
中文翻译:
在经典分子动力学模拟中对化学反应进行建模
提出了一种能够在传统的固定化合价场下将多步反应机制纳入原子分子动力学模拟的算法,并在LAMMPS(大型原子分子大规模并行模拟器)的框架内实现。此扩展称为固定键/反应可以在运行的MD模拟过程中使用反应前和反应后结合模板进行结合拓扑修改,以执行预先指定的反应。首先通过原子间分离识别候选反应物,然后应用广义拓扑匹配算法以确认它们与反应前模板匹配。随后进行拓扑转换以匹配反应后模板,并进行动态松弛以最大程度地减少高能配置。模拟了两个案例研究,即尼龙6,6的缩聚和高交联环氧树脂的形成,以证明该算法的鲁棒性,稳定性和速度。讨论了可以增加其实用性的改进。






































 京公网安备 11010802027423号
京公网安备 11010802027423号